La hepatitis autoinmune (HAI) es una enfermedad hepática crónica y progresiva caracterizada por un cuadro histológico de hepatitis de interfase, hipergammaglobulinemia y autoanticuerpos circulantes. La HIA se da en todos los grupos de edad y afecta con más frecuencia a las mujeres que a los hombres (proporción 3:1). El espectro de la HAI va desde la enfermedad asintomática a la hepatitis aguda o incluso fulminante, pasando por la insuficiencia hepática. Los corticosteroides se utilizan para inducir la remisión. En la terapia de mantenimiento, se prefiere la azatioprina para reducir el riesgo de recidiva durante al menos dos años, preferiblemente cuatro.
La hepatitis autoinmune (HAI) representa entre el 11 y el 23% de todas las hepatopatías crónicas. La incidencia puede estar infravalorada porque la hepatitis vírica es frecuente y cuando existe una hepatitis vírica crónica, cualquier hepatitis autoinmune concurrente puede pasar desapercibida. La incidencia anual en caucásicos es de 0,1-1,9/100.000, y la prevalencia es de 16,9/100.000 [1]. La HIA es responsable del 2,6% de los trasplantes de hígado en Europa [2] y del 4-6% en EE.UU. [3]. Las mujeres se ven afectadas unas tres veces más a menudo. Básicamente, la enfermedad se da en todos los grupos de edad, pero en el 50% de los casos, la enfermedad comienza antes de los 30 años.
Patogénesis
La patogénesis exacta de la HIA sigue sin estar clara. El cuadro clínico refleja una compleja interacción entre la predisposición genética, los factores desencadenantes, los autoantígenos y los mecanismos inmunorreguladores, que en última instancia conducen a un proceso inmunológico (mediado por células, por anticuerpos o en combinación) contra los hepatocitos con el desarrollo de una inflamación crónica que desemboca en cirrosis. Se desconocen los factores desencadenantes exactos; son posibles los agentes infecciosos, medicinales y tóxicos. El mimetismo molecular de antígenos extraños y propios es la explicación más común de la pérdida de autotolerancia [4].
Clínica
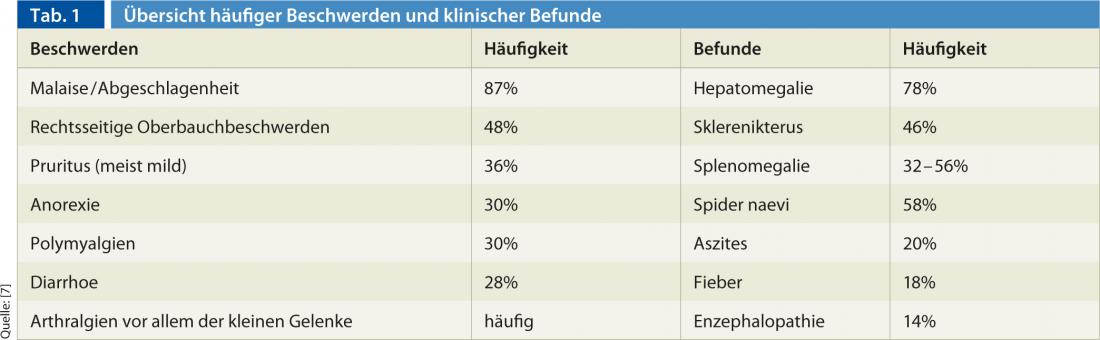
La AIH tiene un aspecto muy heterogéneo y fluctuante. El diagnóstico suele retrasarse porque a menudo sólo se presentan síntomas leves e inespecíficos de una hepatitis aguda autolimitada. La tabla 1 ofrece una visión general de las quejas típicas y los hallazgos clínicos [5].
Existen diferentes formas de manifestación, que se comentan brevemente a continuación:
- Asintomática: El diagnóstico se realiza a partir de un hallazgo incidental de enzimas hepáticas elevadas en pacientes asintomáticos.
- Hepatitis aguda: Aparición aguda en cerca del 40% de los casos. Una anamnesis detallada revela a menudo síntomas inespecíficos anteriores como episodios de malestar, náuseas y artralgia.
- Insuficiencia hepática aguda: En casos raros, presentación inicial como hepatitis fulminante (5%) o descompensación hepática (“enfermedad quemada”) con ascitis (20%), encefalopatía hepática (14%) y varices esofágicas sangrantes (8%).
- Cirrosis hepática: Hasta un 30% de los diagnósticos iniciales como cirrosis hepática o con las complicaciones correspondientes.Una característica típica de la HIA es su asociación con síndromes inmunomediados extrahepáticos como la tiroiditis autoinmune, el vitíligo, la alopecia, la colitis ulcerosa, la artritis reumatoide, la diabetes mellitus y la glomerulonefritis.
División
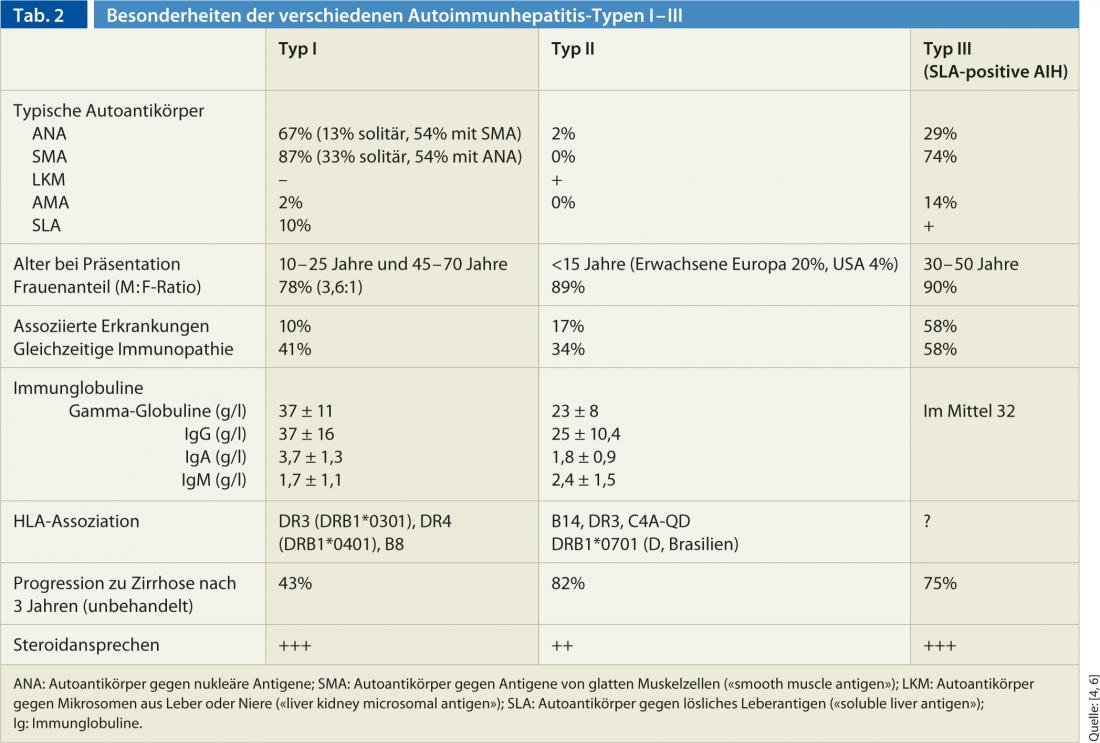
Basándose en la constelación de anticuerpos, la HIA se clasifica en tres tipos diferentes (tipo I-III) (Tabla 2). La HIA de tipo I es la HIA “clásica” y la forma más común en todo el mundo. En comparación, la HIA de tipo II es poco frecuente y afecta principalmente a pacientes pediátricos (niñas/mujeres jóvenes). La aparición de la HIA puede darse en combinación con otras enfermedades autoinmunes del hígado (los llamados “síndromes de solapamiento”). La HIA suele coincidir con la cirrosis biliar primaria AMA-positiva (alrededor del 88%), pero la colangitis esclerosante primaria y la hepatitis vírica también pueden estar presentes al mismo tiempo.
Diagnóstico
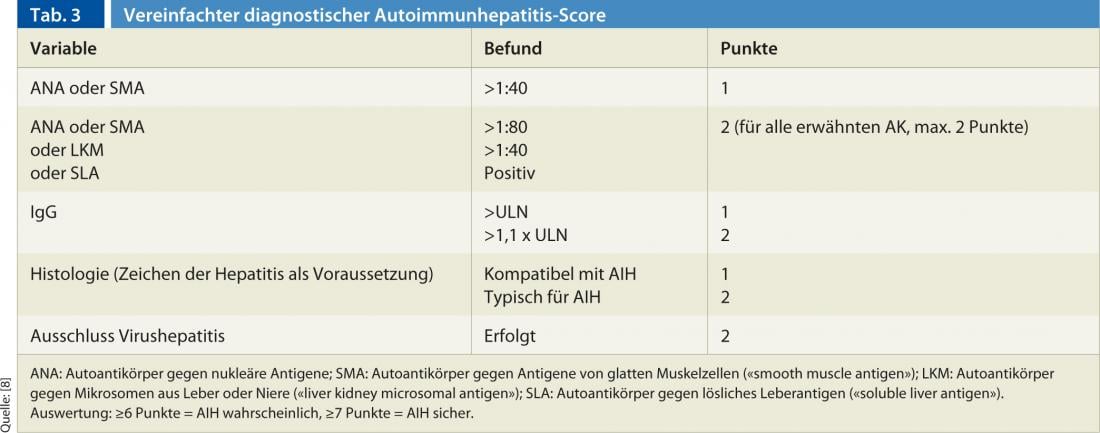
El diagnóstico se basa en una combinación de hallazgos clínicos, histológicos y serológicos, así como en la exclusión de otras hepatopatías crónicas (principalmente hepatitis víricas, hepatitis inducidas por fármacos, hepatopatías hereditarias y metabólicas) [7]. Ningún hallazgo por sí solo es patognomónico de la HIA. Por este motivo, se han establecido varias puntuaciones en el pasado, siendo la recientemente publicada y simplificada de Hennes et al. es fácil de manejar en la práctica clínica diaria [8]. La puntuación distingue entre un diagnóstico definitivo (mín. 7 puntos) y uno probable (mín. 6 puntos) de HIA y se basa en los cuatro criterios siguientes (Tabla 3):
- Presencia de autoanticuerpos
- Inmunoglobulina G (IgG) elevada
- Histología hepática
- Exclusión de hepatitis vírica
La sensibilidad y la especificidad de esta puntuación son elevadas: para un diagnóstico definitivo, los valores son del 81% y el 99%, respectivamente; para un diagnóstico probable, del 88% y el 97% [8].
Laboratorio: Los exámenes químicos de laboratorio suelen mostrar el cuadro típico del daño hepático hepatocelular: Las transaminasas son claramente superiores a los parámetros de colestasis (relación AST: ALP >3). Un cuadro colestásico con hiperbilirrubinemia directa predominante y elevación de la ALP es más raro. Además, si se sospecha de HIA, deben determinarse las gammaglobulinas y los autoanticuerpos contra antígenos nucleares (ANA), contra antígenos de células musculares lisas (“antígeno de músculo liso” [SMA]) y contra microsomas del hígado o del riñón (“antígeno microsómico de hígado y riñón” [LKM]).
Entre los autoanticuerpos adicionales más recientes se incluyen los anticuerpos contra el citosol hepático 1 (LC-1), cuyos títulos se correlacionan con la actividad inflamatoria de la HIA, o los anticuerpos contra el receptor de asialoglicoproteína (ASGPR). Pueden detectarse hasta en el 90% de los casos de HIA de tipo I y tienen importancia pronóstica.
Histología: La histología es esencial para hacer un diagnóstico definitivo, aunque los hallazgos histológicos no sean específicos de la HIA [5]. Aunque la presentación clínica sea aguda, histológicamente suele haber ya signos de hepatopatía crónica. La expresión histológica no se correlaciona con el grado de elevación de los valores hepáticos o de IgG. Así pues, la histología es un parámetro más fiable que los hallazgos de laboratorio para estimar la gravedad de la enfermedad [9].
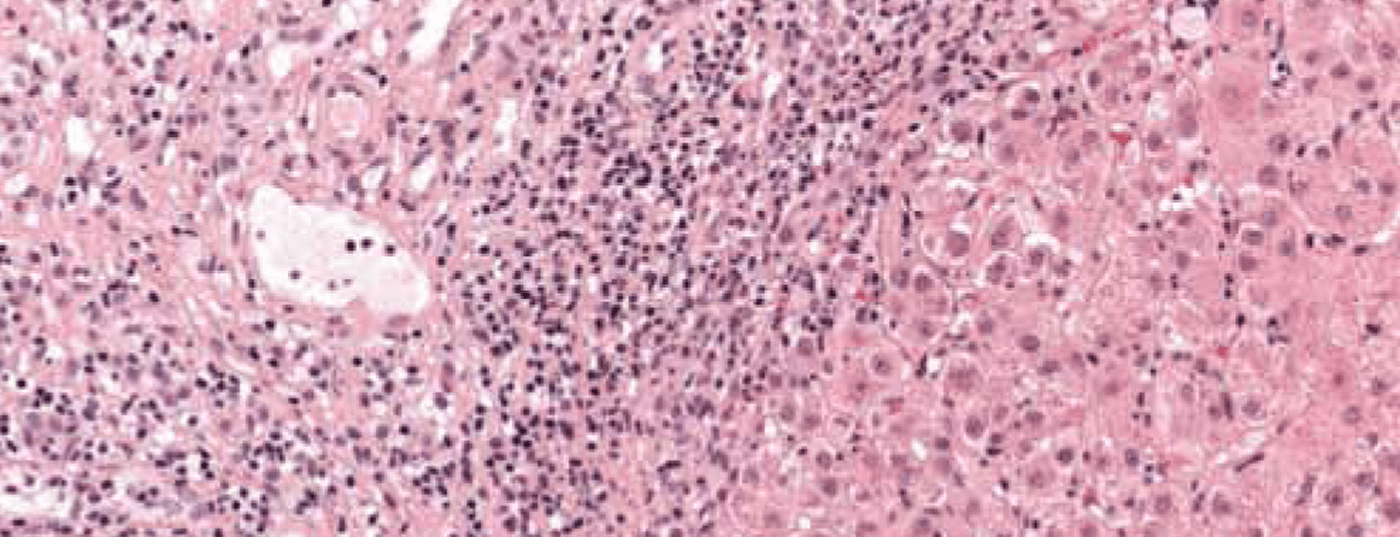
El cuadro de una enfermedad necroinflamatoria crónica es característico: infiltrados portales y periportales (“necrosis fragmentaria”, “hepatitis de interfase”) así como monocíticos (linfoplasmocíticos) (Fig. 1). En la enfermedad grave y avanzada, existe una extensa necrosis en puente y fibrosis. La “hepatitis interfacial” no indica necesariamente un curso progresivo de la enfermedad, mientras que la “necrosis en puente” aumenta la probabilidad de progresión a cirrosis. La especificidad del hallazgo histológico global es del 81% y el valor predictivo positivo del 68%. Por ello, a veces puede resultar difícil diferenciar la hepatitis inducida por fármacos o la hepatitis vírica de la HIA.
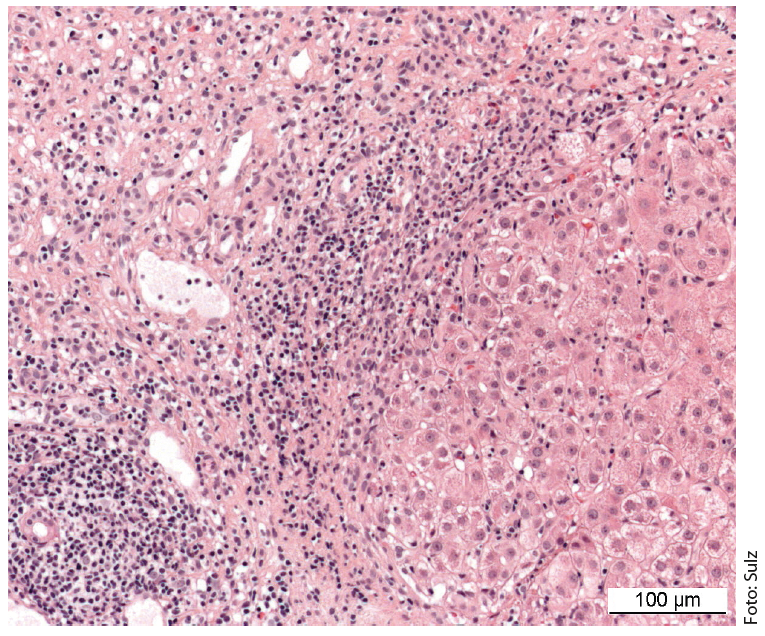
Fig. 1: La histología hepática de una niña de 12 años muestra secciones del campo portal parcialmente fibrosadas con densos infiltrados inflamatorios linfoplasmocitarios y hepatitis de interfase activa. Tinción: hematoxilina-eosina.
Terapia
Indicaciones: Según la Asociación Americana para el Estudio de las Enfermedades Hepáticas (AASLD), existen indicaciones absolutas y relativas para el tratamiento de la HIA (Tabla 4) [3].

Clasificación de la terapia: La terapia puede dividirse en diferentes fases:
- Inducción a la remisión
- Mantenimiento de la remisión
- Disminución o interrupción
- Terapia de una recaída
Objetivo de la terapia: El objetivo de la terapia es la remisión permanente. Clásicamente, la remisión se define como un descenso de la AST <2 veces la norma superior y la normalización de las gammaglobulinas [8]. Sin embargo, estudios recientes indican que la normalización completa de las enzimas hepáticas, la histología y las gammaglobulinas debe ser el objetivo de la terapia, ya que esto ha mejorado el pronóstico de los pacientes tratados [10, 11].
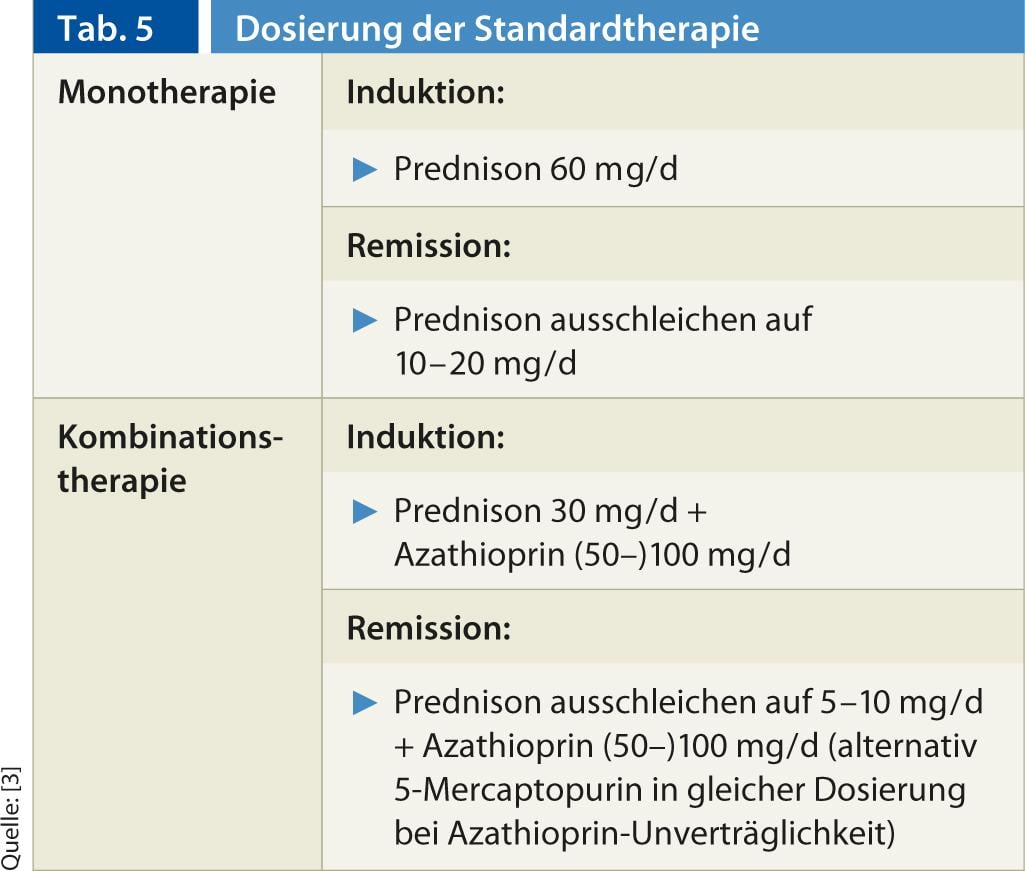
Terapia estándar: Para la terapia inmunosupresora de la HIA, la cooperación entre el médico de atención primaria y un especialista ha demostrado su eficacia. Actualmente, la terapia estándar consiste en monoterapia o terapia combinada con prednis(ol)-ona con/sin azatioprina, que se basa en estudios prospectivos, no aleatorizados, desde los años 60 hasta principios de los 80. Los regímenes de dosificación se mencionan en la Tabla 5. Es importante que la dosis de esteroides no se reduzca demasiado rápido. Debe esperarse la normalización de las transaminasas. En el caso de la terapia combinada esteroides/tiopurina, la azatioprina se añade rápidamente al esteroide. En caso de intolerancia a la azatioprina, es posible cambiar a la 5-mercaptopurina (Puri-Nethol) en la misma dosis. La eficacia de la combinación y la monoterapia es comparable, pero la terapia combinada ahorra esteroides y se asocia a efectos secundarios menos graves de los esteroides (del 66% al <20% durante 18 meses de terapia) [12].
Terapia alternativa con budesonida: En el mayor estudio de terapia controlada realizado hasta la fecha sobre la HIA, se comparó el valor de la budesonida (3 × 3 mg) con el de la prednis(ol)-ona en cada caso en combinación con azatioprina [13]. Ambos brazos del estudio mostraron una tasa relativamente baja de remisiones completas, pero significativamente menos efectos secundarios de los esteroides en el grupo de la budesonida (28% frente a 53%). La budesonida (2 × 3 mg) también parece ser adecuada para el mantenimiento de la remisión.
Sin embargo, la hipertensión portal o la cirrosis constituyen una contraindicación relativa para la budesonida debido a su metabolismo reducido [14].
Importancia de la remisión y la recaída tras una duración suficiente de la terapia: La remisión se consigue en el 65-75% de los casos tras 24 meses de terapia. Alrededor del 20% no logra una remisión completa. La recurrencia (aumento de las transaminasas y/o de los síntomas durante la terapia o durante su disminución o tras la interrupción de la misma) se produce en cerca del 50% de los casos en un plazo de seis meses y en el 80% después de tres años tras la interrupción de la terapia, asociándose con la progresión a cirrosis hepática en casi el 40% y el desarrollo de insuficiencia hepática en el 14%.
La duración de la terapia antes de la interrupción de la inmunosupresión parece ser el principal factor para distinguir a los pacientes en remisión sostenida de los pacientes con recaída. En comparación con la remisión sostenida del 67% con terapia >4 años, esta tasa es sólo del 10% con terapia de 1-2 años [15]. Por lo tanto, se recomienda interrumpir la terapia sólo si se produce una remisión clínica, histológica y bioquímica completa al cabo de dos años, preferiblemente cuatro, como muy pronto. La remisión histológica puede retrasarse hasta ocho meses en comparación con la remisión clínica y bioquímica.
Nuevas opciones terapéuticas (“segunda línea”): En los siguientes casos deben discutirse opciones de tratamiento alternativas:
- Fracaso del tratamiento con la terapia estándar
- Intolerancia a la terapia estándar
- Evitar los efectos secundarios de los esteroides en pacientes de alto riesgo
- Estudios experimentales
Aquí se utilizan, entre otros, el micofenolato, el mofetil (MMF), el tacrolimus o la ciclosporina A.
Trasplante de hígado: Si la terapia fracasa, debe considerarse el trasplante de hígado en una fase temprana. Tras un trasplante exitoso, la supervivencia es buena (supervivencia a 5 años: 91% [16]; supervivencia a 10 años: 75% [cirrosis: 62%]). El riesgo de recidiva en el trasplante es del 20-36% [16] y es aún más frecuente en los niños.
Control y seguimiento de la terapia: Las transaminasas y el nivel de IgG indican el éxito de la terapia y deben determinarse regularmente (de tres a seis meses). La determinación de autoanticuerpos clásicos durante el curso no es útil, ya que el nivel del título no se correlaciona con el grado de actividad. Se recomienda realizar una biopsia hepática antes de interrumpir el tratamiento, ya que una reacción inflamatoria residual en el hígado puede indicar ya un mayor riesgo de recidiva.
En presencia de cirrosis, se recomienda el cribado del carcinoma hepatocelular. Además, los pacientes con HIA deben recibir la vacuna contra la hepatitis A/B, así como las vacunas estándar habituales [17].
Situaciones terapéuticas especiales
Pacientes de riesgo: Los riesgos de efectos secundarios de la terapia son: metabolismo diabético, osteoporosis, hipertensión arterial mal controlada, labilidad emocional o antecedentes positivos de psicosis. Estas situaciones de riesgo no son una contraindicación absoluta, pero debe vigilarse estrechamente a los pacientes y utilizar antes sustancias ahorradoras de esteroides como la azatioprina.
Hepatitis fulminante: Aunque un pequeño estudio no logró demostrar un beneficio relevante de los esteroides en la HIA fulminante, la respuesta a los esteroides en la presentación grave fue del 36-100% en otros estudios. En general, se recomienda la derivación inmediata a un centro de trasplantes en caso de presentación fulminante [18, 19].
Cirrosis: Los pacientes con y sin cirrosis muestran un resultado comparativamente bueno (muerte y trasplante de hígado como criterios de valoración) al cabo de diez años (supervivencia a 10 años >90%), tras lo cual la curva de supervivencia de los pacientes con cirrosis (supervivencia a 20 años <40%) desciende significativamente en comparación con los que no tienen cirrosis (supervivencia a 20 años <80%).
Cirrosis descompensada: Aunque el beneficio del tratamiento no está claro en la cirrosis histológicamente inactiva, la respuesta al tratamiento en la cirrosis descompensada con hepatitis activa puede ser muy buena. En algunos casos, también es posible la regresión de la fibrosis [20].
Previsión
Los pacientes con HIA bajo un tratamiento adecuado tienen una esperanza de vida normal en general con una calidad de vida bien mantenida. Sin embargo, alrededor del ¹⁄3 de los pacientes con HIA presentan ya una cirrosis hepática completa en el momento del primer diagnóstico, con el correspondiente empeoramiento del pronóstico. A excepción de los cursos fulminantes, el trasplante de hígado puede evitarse en casi todos los pacientes no cirróticos y en la mayoría de los pacientes con cirrosis temprana.
Dr. Michael Christian Sulz
PD Dr. med. Tilman J. Gerlach
CONCLUSIÓN PARA LA PRÁCTICA
- (AIH) representa entre el 11 y el 23% de todas las hepatopatías crónicas, aunque su incidencia puede estar infravalorada.
- La patogénesis exacta de la HIA sigue sin estar clara. El cuadro clínico refleja una compleja interacción entre la predisposición genética, los factores desencadenantes, los autoantígenos y los mecanismos inmunorreguladores, que en última instancia da lugar a un proceso inmunológico contra los hepatocitos.
- La HIA tiene una presentación muy heterogénea y fluctuante con diferentes manifestaciones que van desde la asintomática a la hepatitis aguda, pasando por la insuficiencia hepática aguda y la cirrosis hepática.
- El diagnóstico se basa en una combinación de hallazgos clínicos, histológicos y serológicos, así como en la exclusión de otras hepatopatías crónicas. La puntuación simplificada de Hennes et al. es fácil de manejar en la práctica clínica diaria.
- La terapia puede dividirse en diferentes fases como la inducción, el mantenimiento de la remisión, la disminución o interrupción y la terapia de una recaída.
- El objetivo de la terapia es la remisión permanente; la terapia estándar es la monoterapia o la terapia combinada con prednis(ol)on con/sin azatioprina.
Literatura:
- Boberg KM, et al: Scand J Gastroenterol 1998; 33: 99-103.
- Milkiewicz P, et al: Transplantation 1999; 68: 253-256.
- Manns MP, et al: Hepatology 2010; 51: 2193-2213.
- Yeoman AD, et al: Hepatología 2009; 50: 538-545.
- Czaja AJ, Freese DK.: Hepatología 2002; 36: 479-497.
- Sleisinger, Fordtran’s.: Enfermedades gastrointestinales y hepáticas. 8ª ed. Vol.2. Saunders, 2006.
- Krawitt EL.: N Engl J Med 2006; 354: 54-66.
- Hennes EM, et al: Grupo Internacional de Hepatitis Autoinmune. Hepatología 2008; 48: 169-176.
- Czaja AJ, Wolf AM, Baggenstoss AH: Gastroenterología 1981; 80: 687-692.
- Miyake Y, et al: J Hepatol 2005; 43: 951-957.
- Montano-Loza AJ, Carpenter HA, Czaja AJ.: Am J Gastroenterol 2007; 102: 1005-1012.
- Summerskill WH, et al: Gut 1975; 16: 876-883.
- Manns MP, et al: Gastroenterología. 2010; 139: 1198-1206.
- Geier A, et al: World J Gastroenterol 2003; 9: 2681-2685.
- Kanzler S, et al: J Hepatol 2001; 34: 354-355.
- Campsen J, et al: Trasplante de hígado 2008; 14: 1281-1286.
- Gleeson D, Heneghan MA.: Gut 2011; 60: 1611-1629.
- Kessler WR, et al: Clin Gastroenterol Hepatol 2004; 2: 625-631.
- Czaja AJ: Trasplante de hígado 2007; 13: 953-955.
- Dufour JF, DeLellis R, Kaplan MM: Ann Intern Med 1997; 127: 981-985.
PRÁCTICA GP 2013; 5: 10-14