
Los criterios diagnósticos actualmente válidos para la esclerosis múltiple (EM) permiten un diagnóstico muy precoz y específico en la mayoría de los casos. Sin embargo, cuando se sospecha una EM, siempre es importante tener en cuenta los diagnósticos diferenciales más comunes y excluir los “mimetismos”. El diagnóstico de los síndromes similares a la EM tiene consecuencias terapéuticas y, por supuesto, pronósticas. Si surgen dudas sobre el diagnóstico de EM durante el curso de la enfermedad, no debe dudarse en renovar la validación del diagnóstico diferencial.
Hasta la fecha, no existen marcadores biológicos para el diagnóstico definitivo de la esclerosis múltiple (EM). Ningún síntoma neurológico clínico es patognomónico, ya que los focos de EM pueden aparecer en cualquier parte del SNC. El tiempo medio transcurrido entre el primer síntoma, que a menudo no se reconoce, y el diagnóstico sigue siendo de unos dos años [1]. Al ser la enfermedad neurológica más común de la edad adulta joven, puede provocar una discapacidad precoz y la amenaza de una jubilación anticipada. A la vista de los hallazgos fisiopatológicos y del inicio precoz de la terapia que se requiere hoy en día, se trata de una pérdida de tiempo que ya no puede recuperarse.
La segunda revisión de los criterios McDonald permite diagnosticar la EM en el primer episodio y con una RM de una sola sesión, pero aún así sólo con la exclusión más cuidadosa de otros posibles diagnósticos diferenciales (Tablas 1 y 2) [2–5].

En el sentido de un procedimiento según el principio de “diagnosticar con frecuencia cosas comunes”, los diagnósticos diferenciales (DD) que se plantean en nuestras latitudes y en el paciente “típico”, es decir, más joven y con sospecha de EM, se tratarán a continuación de forma superficial.
Edad e historial médico
La distribución por edades de la EM muestra un pico de la enfermedad en torno a los 30 años. La EM en niños y adolescentes y la EM de primera aparición después de los 45 años son menos frecuentes. Sin embargo, existen casos individuales bien documentados de EM que se manifestaron por primera vez en la primera década de vida, pero también los que se manifestaron en la séptima década.
Realizar una anamnesis precisa es esencial y suele ser suficiente para establecer una sospecha diagnóstica de EM. La anamnesis debe incluir preguntas sobre cualquier episodio anterior de déficit neurológico que pueda proporcionar pistas sobre recaídas pasadas que puedan haber sido malinterpretadas previamente. También es importante preguntar sobre otras enfermedades autoinmunes en el paciente o en miembros de su familia. Las preguntas sobre quejas y síntomas en el ámbito de la vejiga, el recto y la función sexual son importantes. También deben buscarse síntomas ocultos como fatiga, problemas de concentración, depresión y dolor (disfunción autonómica) [3–5]. Los primeros síntomas comunes de la EM son:
- Trastornos de la sensibilidad como formicación, sensación de pelusilla, hormigueo (>30%)
- Trastornos visuales con visión borrosa, nublada o nebulosa causados por una neuritis óptica unilateral (aprox. 16%)
- Trastornos de la marcha con debilidad frecuente de las piernas dependiente de la carga e inestabilidad de la marcha (aprox. 5%).
Otros síntomas son: Mareo (30-50% de los casos), nistagmo (2-4%), temblor intencional, disfunción vesical e intestinal, disfunción sexual, visión doble, parestesia facial, neuralgia del trigémino, ataxia, disartria, fatiga (sobre todo en la recaída), trastornos del sueño, depresión, euforia, deterioro cognitivo (34-65%), demencia (5%), convulsiones cerebrales (2-3%) y signo de Lhermitte positivo.
Diagnóstico diferencial de la EM
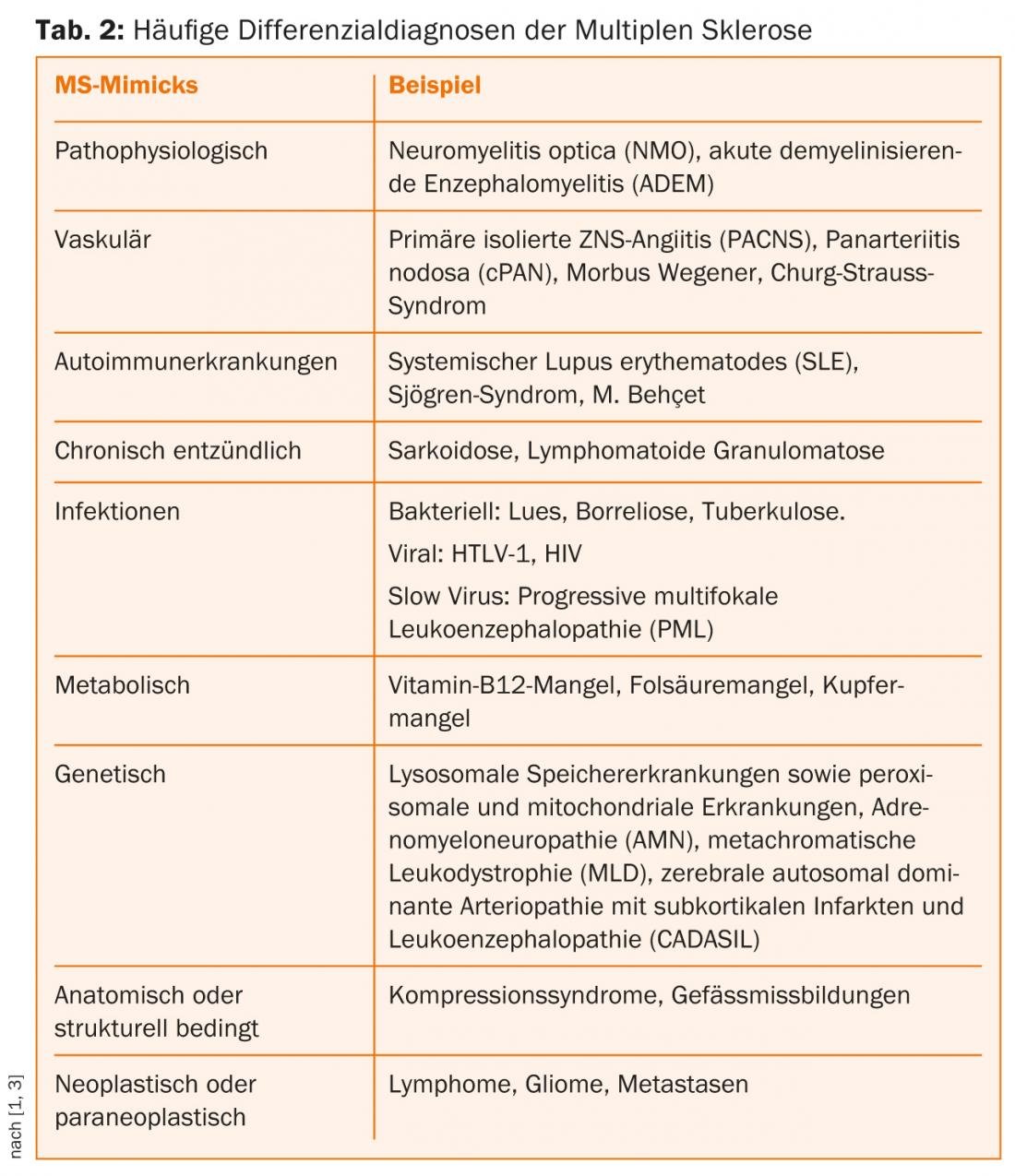
La riqueza de formas de los síntomas de la EM hace que sea tan difícil diferenciarlos de toda una serie de afecciones cerebroespinales. Otras enfermedades crónicas inflamatorias, metabólicas, vasculares, neoplásicas y/o mediadas por patógenos pueden presentarse con síntomas similares (Tab. 2) [3–5].
Imitaciones fisiopatológicas
La EM es una enfermedad inflamatoria crónica inmunomediada del sistema nervioso central que histopatológicamente conduce a diversos grados de daño axonal y desmielinización. Existen otras enfermedades autoinmunes que afectan al sistema nervioso central y que son difíciles de distinguir de la EM debido a la variedad de síntomas.
La neuromielitis óptica (NMO) es también una enfermedad inflamatoria crónica inmunomediada del SNC. Las “enfermedades del espectro de la NMO” (NMOSD) incluyen la mielitis transversa extensa longitudinal aislada (LETM), la neuritis óptica aislada monofásica o recurrente y distintas formas de encefalitis del tronco encefálico. La NMO muestra una clara preferencia por el sexo femenino (hasta 9:1 dependiendo de la cohorte) y se manifiesta de media unos diez años más tarde (4ª década de la vida) que la EM. Debido a la detección de anticuerpos contra la acuaporina-4 (AQP4-Ak), que se dirigen contra los canales de agua expresados en los astrocitos, la NMO se clasificó como una de las denominadas enfermedades autoinmunes mediadas por células B. La detección serológica de AQP4-Ak y los aumentos del recuento celular predominantemente linfocítico de >20 a 50 µl en el LCR no son infrecuentes en la NMO en el contexto de una recaída aguda. Además, sólo se encuentran bandas oligoclonales positivas en alrededor del 15-30% de los pacientes con NMO. En comparación con la EM, las recaídas en la NMO suelen ser más graves y conducen más rápidamente a discapacidades permanentes debido a una remisión en su mayoría incompleta [5].
La encefalomielitis desmielinizante aguda (ADEM) y su variante máxima, la leucoencefalitis hemorrágica aguda (AHLE), son enfermedades desmielinizantes inflamatorias raras del SNC. A diferencia de la EM, el ADEM es más frecuente en la infancia que en los adultos y se produce principalmente en estrecha relación temporal con infecciones o vacunaciones. No existen criterios diagnósticos claramente definidos para el ADEM. Los raros casos en adultos suponen un reto [5].

Angiitis y colagenosis
La angiitis primaria aislada del SNC (PACNS) es una enfermedad autoinmune muy poco frecuente que se caracteriza por el daño inflamatorio de los vasos cerebrales de pequeño y mediano calibre. Puede considerarse un diagnóstico diferencial en presencia de síntomas multifocales o difusos del SNC con un curso progresivo o remitente-recurrente. La resonancia magnética y la angiografía de sustracción digital proporcionan pistas. Si los cambios vasculíticos están presentes fuera del SNC, se excluye el PACNS [5].
Las vasculitis sistémicas (primarias) se clasifican según el tamaño de los vasos afectados y su constelación histológica de hallazgos. La panarteritis nodosa (cPAN), la enfermedad de Wegener y el síndrome de Churg-Strauss son posibles diagnósticos diferenciales de la EM. Se caracterizan por una inflamación granulomatosa y necrosante de los pequeños vasos y la presencia de anticuerpos antinucleares (ANCA).
Las enfermedades autoinmunes como el lupus eritematoso sistémico (LES), el síndrome de Sjögren o la enfermedad de Behçet rara vez pueden causar síntomas neurológicos debidos a la vasculitis. En principio, las hemorragias y los infartos isquémicos en la zona de suministro de las áreas vasculares afectadas pueden complicar el curso de todas las enfermedades autoinmunes inflamatorias crónicas y provocar síntomas neurológicos graves en el SNP y el SNC (vasculitis secundarias).
La enfermedad de Behçet es una vasculitis sistémica y afecta predominantemente a hombres de la región mediterránea. El pico de la enfermedad se sitúa más allá de la tercera década de vida, y la primera manifestación rara vez se produce en la adolescencia. En los afectados se encuentra una asociación sorprendentemente frecuente con el antígeno HLA-B51. Son típicas las ulceraciones orales y genitales recurrentes y los cambios cutáneos en forma de eflorescencias pustulopapulares estériles y eritema nodoso. La afectación ocular es típica. En raras ocasiones, se produce una manifestación parenquimatosa del SNC (meningoencefalitis), que afecta al tronco encefálico en cerca del 50% de los casos. El aspecto neurológico varía en función de la zona del cerebro afectada o de las estructuras vasculares afectadas y puede ser tanto recidivante como crónico progresivo. El Neuro-Behçet se detecta con IRM y diagnósticos de LCR [5].
En enfermedades granulomatosas como la neurosarcoidosis, la afectación del SNC por sí sola es extremadamente rara; la coinfectación se da en aproximadamente el 5% de los pacientes con sarcoidosis. En las manifestaciones del SNC, la afectación principalmente unilateral de los nervios craneales (nervio facial, nervio óptico, nervio acústico) es el evento inicial más común. El aspecto clínico posterior depende esencialmente de la localización y extensión de los granulomas en el SNC.
Causas infecciosas
Mientras que las neurolúes eran la DD más común de la EM en 1925, ahora son raras y las nuevas causas infecciosas son más importantes. Los síntomas neurológicos de la lúes causada por el Treponema pallidum pueden describirse como meningitis precoz (estadio 2), lúes cerebroespinal con arteritis cerebral concomitante (estadio 3) y parálisis progresiva/tabes dorsal (estadio 4) hacer acto de presencia. Especialmente en la fase 3, los síntomas varían mucho. Los focos desmielinizantes en el cerebro y la médula espinal caracterizan el estadio de tabes dorsal [5].
En cuanto al diagnóstico diferencial, debe tenerse en cuenta la neuroborreliosis, sobre todo en las regiones con una alta tasa de infestación de garrapatas con Borrelia burgdorferi, aunque su frecuencia en las personas infectadas con Borrelia sea baja, del 0,3 al 1,4%. Se distinguen tres estadios, en el que el estadio 1, el eritema migrans local, aún no muestra ningún síntoma neurológico. Si no se trata, puede desarrollarse una infección diseminada (estadio 2) al cabo de semanas o meses, que afecta a varios sistemas orgánicos y, por tanto, también al SNC y al SNP. La neuroborreliosis aguda se caracteriza por un dolor radicular segmentario, a menudo resistente a los analgésicos, sin signos meníngeos de irritación. En el curso de la enfermedad, más del 50% de los pacientes desarrollan irritaciones sensoriales, paresia de las extremidades y déficits de los nervios craneales (nervio facial, nervio abducens). Después de meses o años, pueden desarrollarse síntomas en el sentido de una neuroborreliosis crónica progresiva (estadio 3: mielitis, miositis, vasculitis y polineuropatía). La IRM desempeña un papel secundario en la neuroborreliosis; la evidencia de cambios inflamatorios en el LCR y la producción intratecal de anticuerpos específicos de Borrelia son concluyentes en este caso.
Entre los virus, cabe mencionar el virus de la leucemia de células T humanas 1 (HTLV-1), un retrovirus patógeno humano que infecta predominantemente a las células T CD4+ y, en casos muy raros, causa leucemia de células T y/o provoca síntomas neurológicos. La paraparesia espástica tropical es una enfermedad muy rara en nuestras latitudes, que tiene principalmente un curso espinal. La infección por VIH también puede causar encefalomielitis en sus fases avanzadas.
Causas metabólicas
La mielosis funicular es una enfermedad por deficiencia de vitamina B12 que provoca la desmielinización del rombencéfalo, el cerebro lateral cerebeloso y los tractos piramidales, especialmente en la médula cervical y torácica. De forma similar a la deficiencia de vitamina B12, los trastornos del metabolismo del ácido fólico pueden dar lugar a una variedad de síntomas en diferentes grados de gravedad. Los trastornos neurológicos incluyen convulsiones, retrasos en el desarrollo y síntomas mielopáticos similares a los de la mielosis funicular. Las enfermedades por carencia de vitaminas son raras en nuestras latitudes (alcohol, malnutrición). Aún más raros son los trastornos en la utilización de las vitaminas debidos a enfermedades genéticas.
Causas genéticas
En raras ocasiones, las enfermedades monogénicas también pueden presentar similitudes fenotípicas con la manifestación clínica de la EM. Estas enfermedades hereditarias incluyen enfermedades de almacenamiento lisosómico, así como enfermedades peroxisomales y mitocondriales como la atrofia óptica de Leber. Dado que en la mayoría de los casos las manifestaciones clínico-neurológicas se producen ya en la infancia y la niñez, se recurre a los neuropediatras.
La adrenoleucodistrofia ligada al cromosoma X (ALD) es una enfermedad de almacenamiento peroxisomal caracterizada por una degradación deficiente de los ácidos grasos de cadena larga. También suele comenzar en la infancia, pero también existe una forma adulta muy poco frecuente, la adrenomieloneuropatía (AMN: paraparesia espástica, insuficiencia suprarrenal).
En la forma adulta de la leucodistrofia metacromática (LDM ), una de las 30 enfermedades de almacenamiento lisosómico diferentes, pueden aparecer trastornos de la marcha espástica, incontinencia y, en el curso posterior, más raramente atrofia óptica y trastornos del movimiento ocular, además de los síntomas neuropsicológicos que se dan con frecuencia. El factor decisivo en la diferenciación clínica de la EM en la ELM es la afectación del sistema nervioso periférico en forma de una polineuropatía sensitivomotora mayoritariamente simétrica y acentuada distalmente.
Paraneoplasias y neoplasias
Los pacientes con síntomas neurológicos suelen tener mucho miedo de padecer un tumor cerebral o medular o metástasis. Un curso recidivante con tendencias regresivas habla en su contra. El dolor de cabeza, las náuseas matutinas, los vómitos, los cambios de personalidad, el enlentecimiento y los ataques epilépticos pueden producirse tanto en lesiones malignas como benignas que ocupan espacio intracraneal, pero son poco frecuentes en la EM.
¿Sospecha de un diagnóstico erróneo?
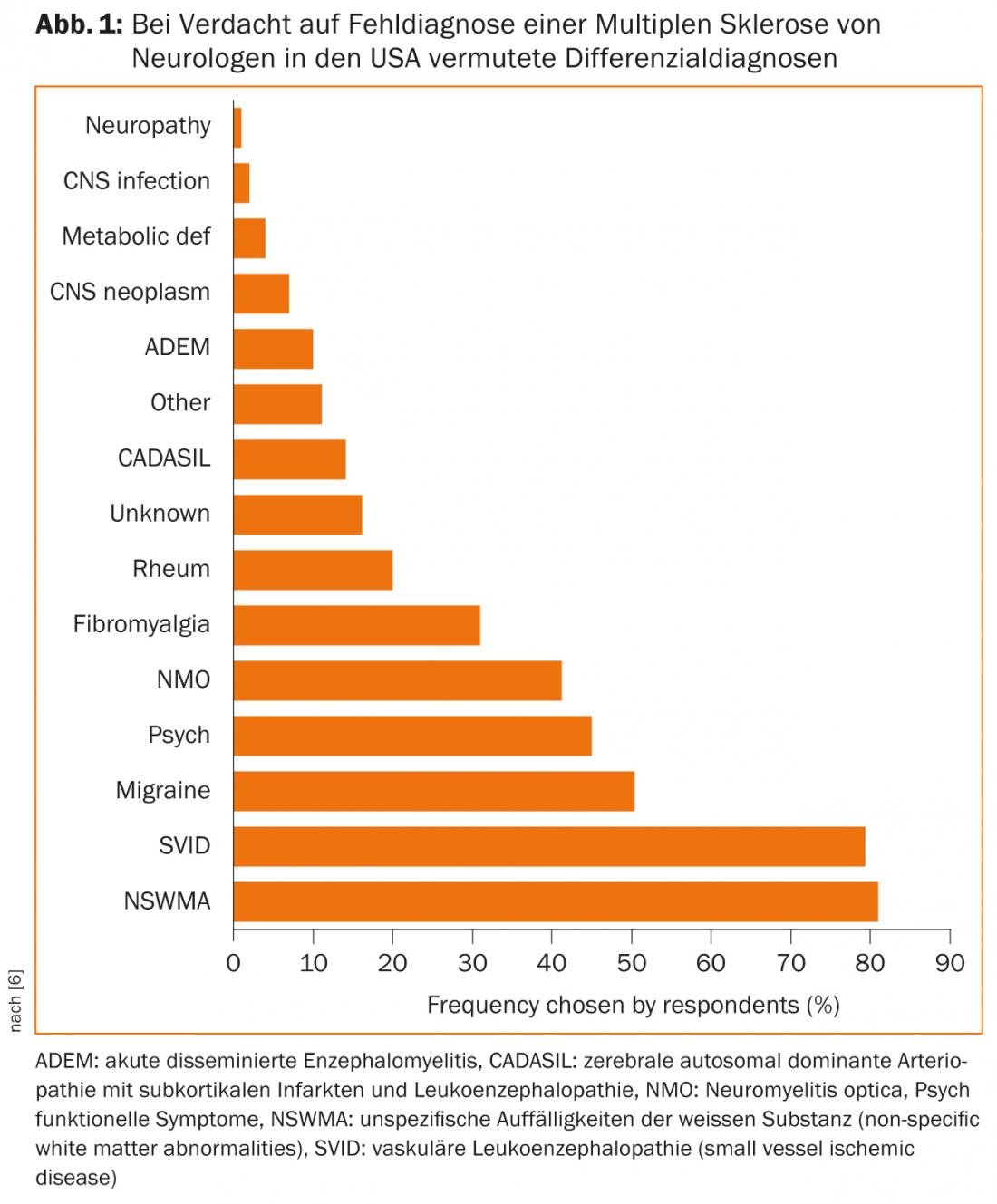
En los casos con un curso atípico, falta de respuesta a las opciones de tratamiento actuales o síntomas fulminantes, es urgente revisar el diagnóstico diferencial y, si es necesario, consultar con expertos de otras disciplinas como reumatólogos e infectólogos. Una encuesta realizada entre neurólogos estadounidenses muestra qué DD se sospechan a menudo cuando se sospecha un diagnóstico erróneo (Fig. 1 ) [6].
Prof. Adam Czaplinski, MD
Literatura:
- Fernández O, et al: Características de la esclerosis múltiple al inicio y retraso del diagnóstico y tratamiento en España (Estudio Novo). J Neurol 2010; 257(9): 1500-1507.
- Polman CH, et al: Criterios diagnósticos de la esclerosis múltiple: revisiones de 2010 de los criterios McDonald. Ann Neurol 2011; 69: 292-302.
- Marcus JF, et al: Actualizaciones sobre el síndrome clínicamente aislado y los criterios diagnósticos de la esclerosis múltiple. Neurohospitalist 2013; 3(2): 65-80.
- www.awmf.de: Esclerosis múltiple Directriz DGN Número de registro AWMF: 030/050.
- Décard BF, et al: Diagnóstico diferencial de la esclerosis múltiple (EM) – Mímicos de la EM. Act Neurol 2012; 39: 83-99.
- Solomon AJ, et al: Undiagnosing MS: The Challenge of misdiagnosis in MS. Neurología 2012; 78; 1986-1991.
InFo Neurología y Psiquiatría 2013; 11(6)









