Siendo el trastorno hereditario de la coagulación más común, el síndrome de Von Willebrand afecta aproximadamente a una de cada mil personas. Con las nuevas posibilidades de diagnóstico y los descubrimientos en el campo de la genética molecular, cada vez se plantea más la cuestión de la importancia clínica de los análisis genéticos. Éste fue también un tema muy debatido en la 63ª Reunión Anual de la Sociedad Americana de Hematología (ASH).
Pruebas genéticas en el síndrome de von Willebrand (Enfermedad de von Willebrand, vWD) es como el amor, todo el mundo habla de él, pero nadie lo entiende exactamente – con estas palabras Emmanuel Favaloro, un reconocido investigador australiano de la Institutos de Patología Clínica e Investigación Médica en Hospital de Westmead en Sydney, su conferencia sobre Reunión anual de la ASH 2021. Y, de hecho, existe cierta incertidumbre sobre la mejor manera de proceder. Aunque el análisis genético no tiene sentido en la mayoría de los casos de síndrome de von Willebrand de tipo 1, se recomienda para los pacientes de tipo 2 y 3 en las nuevas directrices publicadas en 2021 [2].
Síndrome de von Willebrand: Conceptos básicos
El síndrome de Von Willebrand se describió por primera vez en 1926 como “pseudohemofilia” en una joven que se desangró durante una de sus primeras menstruaciones. El descubridor – nomen est omen – es el Dr. Erik von Willebrand. La enfermedad hereditaria autosómica se define por anomalías cuantitativas o cualitativas del factor von Willebrand (FvW), la prevalencia sintomática es de aproximadamente 1/1000. Aunque hombres y mujeres se ven afectados formalmente por igual, las mujeres son diagnosticadas dos o tres veces más a menudo debido a las complicaciones ginecológicas. En la mayoría de los casos, el síndrome de von Willebrand se manifiesta como un aumento de las hemorragias mucosas; en los casos graves, también pueden producirse hemorragias musculoesqueléticas.
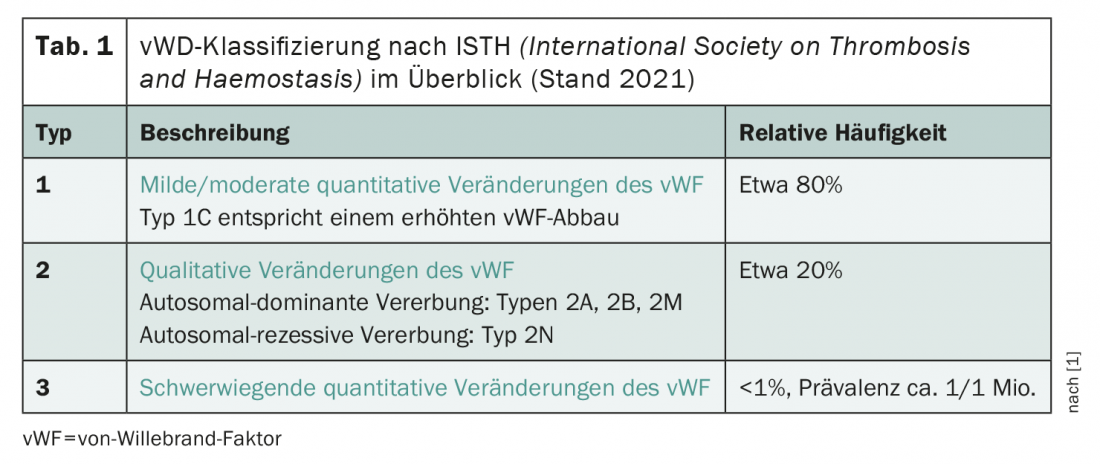
Según la clasificación recientemente revisada de la Sociedad Internacional de Trombosis y Hemostasia (ISTH), el cuadro clínico se divide a grandes rasgos en tres tipos (tab. 1) . En principio, se distingue entre cualitativo (tipo 2) y cambios cuantitativos (tipos 1 y 3) del FvW. En 2021, se añadió a la clasificación el subtipo 1C, que se define por una mayor degradación del FvW. El tipo 1 El síndrome de von Willebrand -es decir, cambios cuantitativos de leves a moderados en el FvW- es, con diferencia, el más frecuentemente diagnosticado, seguido del tipo 2. La ausencia casi completa de FvW en el síndrome de von Willebrand de tipo 3 es extremadamente rara y afecta aproximadamente a una de cada millón de personas.
Además de la terapia sintomática con fármacos antifibrinolíticos como el Cyklokapron y la píldora en pacientes femeninas, también existen opciones de tratamiento dirigidas directamente al FvW. Éstas consisten en desmopresina (DDAVP), que promueve la liberación de FvW de las células endoteliales, y FvW recombinante o derivado del plasma sanguíneo. En principio, la terapia para todos los tipos de síndrome de von Willebrand es similar y depende de la gravedad. En los casos más graves, suele ser necesario sustituir el FvW, ya que el efecto del DDAVP disminuye rápidamente tras un breve periodo de tratamiento.
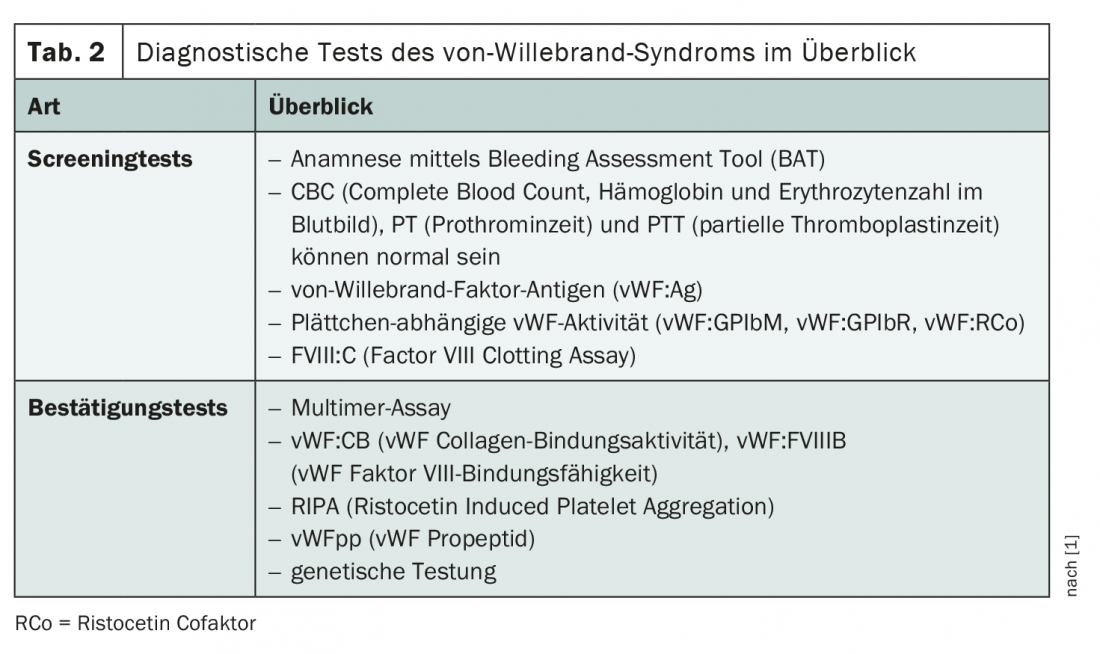
Para realizar el diagnóstico se utilizan varias pruebas de detección y de confirmación (tab. 2) . El historial médico también desempeña un papel decisivo. Dado que el hemograma, el tiempo de protrombina y el tiempo parcial de tromboplastina pueden ser normales, son necesarios procedimientos de cribado más específicos. Suele realizarse un panel de tres herramientas de diagnóstico para detectar antígenos del FvW, actividad del FvW dependiente de las plaquetas y actividad del factor VIII. Si la sospecha de síndrome de von Willebrand se confirma durante estas investigaciones, se utilizan pruebas adicionales para confirmar e identificar el subtipo de vWD.
Pruebas genéticas controvertidas
En el contexto de las pruebas complementarias, el análisis genético también desempeña un papel hasta ahora controvertido. Dado que la terapia no varía entre los subtipos y que se dispone de métodos alternativos más favorables para clasificar la enfermedad, se plantea la cuestión de la utilidad de la caracterización genética. Su disponibilidad varía, pero va en aumento. No deben descuidarse los posibles efectos negativos sobre las personas afectadas, ya que las pruebas genéticas pueden acarrear problemas de seguro y estigmatización, por ejemplo. La conclusión es que el análisis genético del subtipo más común de vWD, el tipo 1, no suele ser útil, según los expertos reunidos en la Reunión Anual de la ASH, entre otras cosas, porque sólo puede detectarse una variante genética correspondiente en aproximadamente el 65% de los casos y esto no tiene ninguna repercusión en el tratamiento.
Aunque se ha avanzado mucho en la caracterización genética de la enfermedad, que se logró por primera vez en 1984, la base de datos sigue llena de errores y lagunas. Por ejemplo, existen algunas denominadas “variantes de significado clínico incierto” y ciertas alteraciones genéticas aún no han podido asignarse claramente a sus fenotipos correspondientes. No obstante, una ventaja de las pruebas genéticas es su viabilidad centralizada y, por tanto, las buenas posibilidades de estandarización. El gen del FvW está situado en el brazo corto del cromosoma 12.
¿Cuándo es útil el análisis genético?
A pesar de todas las reservas, existen algunas situaciones en las que las pruebas genéticas en el síndrome de von Willebrand ya son muy valoradas hoy en día. Especialmente en el caso de los subtipos 2B, 2N y 3 de la vWD, puede proporcionar información adicional importante y clínicamente relevante, por un lado para la diferenciación de otros trastornos de la coagulación y, por otro, para el asesoramiento genético y la evaluación del riesgo terapéutico. Pueden detectarse mutaciones en cerca del 90% de los casos de síndrome de von Willebrand de tipo 2, y en cerca del 85% de los casos de tipo 3. Con respecto al procedimiento diagnóstico óptimo, incluidos los análisis genéticos, en 2021 se publicaron nuevas directrices [2].
Éstos recomiendan la realización de pruebas genéticas en caso de sospecha de vWD de los subtipos 2A, 2B, así como 2N [2]. La cuestión principal aquí es la demarcación del llamado “De tipo plaquetario síndrome de von Willebrand” (también conocido como “pseudotipo”) y la hemofilia A. Esto se debe a que el síndrome de von Willebrand de tipo 2B -que se caracteriza típicamente por una mayor afinidad por las plaquetas, un análisis multimérico conspicuo y trombocitopenia- se parece mucho a la De tipo plaquetario. Sin embargo, mientras que en este último la mutación desencadenante se localiza en el gen de las plaquetas, en el síndrome de von Willebrand tipo 2B se localiza en el gen del FvW. Esto tiene importantes implicaciones para el tratamiento: el síndrome de von Willebrand “real” se trata mediante la sustitución del FvW, el pseudotipo mediante la administración de plaquetas. El tipo plaquetario se hereda de forma autosómica dominante. La causa es una mutación de ganancia de función dela glicoproteína que se une al FvW. Esto conduce a una interacción excesiva e innecesaria entre las plaquetas y el FvW, con un consumo consecutivo de ambos componentes, y a un cuadro clínico que a menudo se asemeja al tipo más común de síndrome de von Willebrand “verdadero”. 2B está confuso. Se estima que alrededor del 15% de los tipos 2B diagnostica en verdad un síndrome de von Willebrand de tipo plaquetario [3].
También existe un diagnóstico diferencial importante para el síndrome de von Willebrand de tipo 2N (“Normandía”): la hemofilia A. En este subtipo, los niveles del factor VIII son clásicamente más bajos que los del FvW – una constelación que sólo se encuentra en el síndrome de von Willebrand en el caso del tipo 2N se produce y se asocia a la hemofilia A puede confundirse. Así, las nuevas directrices recomiendan que, en caso de sospecha de tipo 2N vWD las pruebas genéticas específicas y/o la determinación de la capacidad de unión al factor VIII del vWF [2]. Sólo identificando correctamente la enfermedad subyacente puede garantizarse una terapia adecuada, ya sea mediante la sustitución del factor VIII o del FvW.
A diferencia del síndrome de von Willebrand de tipo 2, el diagnóstico del tipo 3 suelen aclararse antes de las pruebas genéticas. No obstante, puede tener sentido en esta forma rara y autosómica recesiva de la enfermedad, aunque la localización de la mutación desencadenante varía más que en los tipos genéticamente más claramente caracterizados. 2A, B y N. Por un lado, el análisis genético puede desempeñar un papel importante en el asesoramiento genético de las personas afectadas y sus familiares y, por otro, proporciona información valiosa para la evaluación del riesgo de una terapia con FvW exógeno. Así, las grandes deleciones predisponen a la formación de aloanticuerpos durante la terapia. Los pacientes con mutaciones en el propéptido del FvW también parecen tener un mayor riesgo de hemorragia que aquellos con mutaciones en otras localizaciones.
La conclusión es que el beneficio de las pruebas genéticas en el tipo 1 síndrome de von Willebrand – y por tanto en más del 70% de todos los afectados – es actualmente cuestionable. Además de la correlación genotipo-fenotipo insuficientemente caracterizada, también falta la consecuencia terapéutica de estos análisis. Para los tipos 2 y 3, por otro lado, el análisis genético puede ser bastante útil y ya es muy valorado en el diagnóstico diferencial, la evaluación de riesgos y el asesoramiento genético.
Congreso: 63ª Conferencia Anual de la ASH
Fuente/Literatura:
- Sesión destacada “Pruebas genéticas para la enfermedad de von Willebrand”, Paula James y Emmanuel Favaloro, 13.12.2021, 63ª Reunión Anual de la ASH, Atlanta, EE.UU.
- James PD, et al: Directrices ASH ISTH NHF WFH 2021 sobre el diagnóstico de la enfermedad de von Willebrand. Blood Adv. 2021; 5(1): 280-300.
- Othman M: Enfermedad de von Willebrand de tipo plaquetario: un trastorno hemorrágico poco frecuente, a menudo mal diagnosticado e infradiagnosticado. Semin Thromb Hemost. 2011; 37(5): 464-469.
InFo ONCOLOGÍA Y HEMATOLOGÍA 2022; 10(1): 38-40