
A finales de 2013 se celebró el 55º congreso anual de la Sociedad Americana de Hematología. Más de 22.000 participantes acudieron a Nueva Orleans, o “The Big Easy”, como se conoce a la ciudad. Los interesados pueden encontrar el programa completo con extractos de las ponencias en www.hematology.org.
Trasplante haploidéntico de células madre
(mb) El trasplante haploidéntico de células madre hematopoyéticas HLA es una opción eficaz para los pacientes que necesitan un trasplante pero no pueden encontrar un donante con tipos HLA completamente compatibles. Este es el resumen de la doctora Alice Bertaina, del Hospital Infantil Bambino Gesu de Roma. Dicho tratamiento suele asociarse a un mayor riesgo de infección y recaída en comparación con un trasplante de células madre de un donante totalmente compatible. En el método mostrado por Bertaina, las células T alfa/beta+ así como las células B CD19+ se eliminan selectivamente del injerto del donante. Al mismo tiempo, se conservan células inmunoactivas como las células asesinas naturales y las células T gamma/delta+. Un total de 45 niños con leucemia aguda -con edades comprendidas entre 0,9 y 17,9 años durante el trasplante- fueron tratados con células madre de uno de sus progenitores (35 pacientes con LLA y 10 con LMA). El tiempo medio para alcanzar un recuento absoluto de neutrófilos de >0,5 × 109/l y un recuento de plaquetas de >50 × 109/l fue de 13 (9-18) y 11 días (8-20), respectivamente. Ningún niño desarrolló una reacción aguda de injerto-huésped (EICH) en el intestino o el hígado. Se produjo una EICH leve en la piel del 29%. Tras un seguimiento medio de once meses (entre 2 y 30), la estimación de Kaplan-Meier a dos años fue de una tasa de supervivencia libre de leucemia del 75% (IC 95%: 57-86). Este valor fue del 73% (IC del 95%: 52-85) para los pacientes con LLA. “Nuestros resultados con células madre de donantes haploidénticos mostraron tasas de éxito porcentuales coherentes con las de un trasplante de compatibilidad total”, explicó el Dr. Bertaina. “Esto hará que este tratamiento que salva vidas esté disponible para un número mucho mayor de pacientes sin un donante compatible perfecto”.
Receptor quimérico de antígenos (CAR)
Varios investigadores presentaron otro tour de force técnico: una terapia con linfocitos T modificados con CAR para enfermedades oncohematológicas. El receptor de antígeno quimérico (CAR) es una cadena de ADN construida artificialmente que se inserta en las células T de un paciente mediante vectores (conocidos por la genética molecular). Estas “nuevas” células combinan la especificidad de los anticuerpos con el efecto citotóxico de las células T.
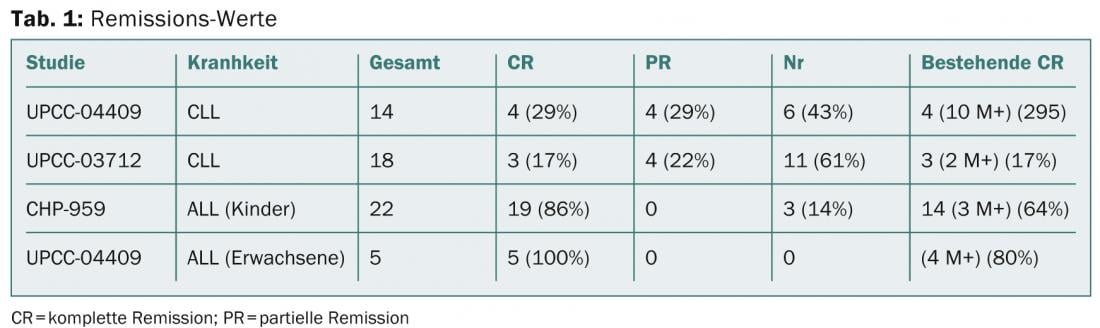
Michael Kalos, de la Facultad de Medicina Perelman de la Universidad de Pensilvania, en Filadelfia, presentó los resultados de estudios en la enfermedad de LLC y LLA avanzada en recaída o refractaria (Porter et al NEJM 2011; Kalos et al Sci Trans Med 2011, Grupp et al NEJM 2013, resumen 67). Se trató a un total de 32 pacientes con LLC muy pretratados, de los que ocho obtuvieron una remisión parcial y siete una remisión completa. Los resultados pueden consultarse en la tabla 1.
CAR para el linfoma de células B
Que los pacientes con linfoma mediastínico primario de células B (PMBCL) y linfoma difuso de células B grandes (DLBCL) refractarios a la quimioterapia también pueden beneficiarse de la terapia CAR anti-CD19 lo demuestra el informe del doctor James Kochenderfer, del Instituto Nacional del Cáncer del Instituto Nacional de Salud en Bethesda. Los investigadores trataron a 20 pacientes con un total de 23 infusiones de células T. Ya se ha informado de los nueve primeros tratamientos con células CAR-T (Kochenderfer et al. en Blood 2010 y Blood 2012). En la ASH, Kochenderfer presentó ahora los resultados aún no comunicados de 14 pacientes. Dado que la quimioterapia previa dejó claro que la actividad de las células T transferidas adoptivamente mejora, los pacientes reciben ciclofosfamida más fludarabina durante cinco días para una infusión de células T CAR anti-CD19. Cinco pacientes alcanzaron la remisión completa (RC) y seis la remisión parcial (RP).
“Este es el primer informe de un tratamiento con éxito de PMBCL y DLBCL refractarios a la quimioterapia con células CAR T anti-CD19”, dijo Kochendorfer. “Nuestros datos son los primeros en sugerir el potencial de este enfoque para pacientes con linfomas agresivos que han sido casi intratables”.
Zevalin y rituximab en comparación
Las terapias de consolidación con Zevalin (90Y ibritumomab-tiuxetan) y rituximab mostraron un beneficio significativo en la supervivencia libre de progresión (SLP). Un estudio español de fase II aclaró que la terapia de consolidación con rituximab es superior a la consolidación con Zevalin en pacientes con linfoma folicular que responden al R-CHOP. De junio de 2008 a julio de 2010, se incluyeron 146 pacientes (66 hombres y 80 mujeres con una edad media de 55 años) de 25 instituciones españolas. El Dr. Armando López-Guillermo, del Hospital Clinic de Barcelona, presentó los resultados preliminares. Tras un seguimiento medio de 37 meses desde la aleatorización (entre 26 y 56), 31 pacientes mostraron progresión avanzada o recidiva. La SLP a 36 meses fue del 64% (IC 95%: 52-76) para los pacientes del grupo de Zevalin y del 86% (IC 95%: 77-95) para los pacientes del grupo de rituximab (p=0,01; HR 0,38; IC 95%: 0,170,83). Durante el periodo de mantenimiento, se produjo neutropenia (grado 3-4) en seis de los 63 pacientes del grupo de Zevalin y trombocitopenia (3-4) en cinco de ellos. Para los pacientes del grupo de rituximab, fue uno o ninguno de los 61 pacientes. Cinco pacientes fallecieron durante el seguimiento debido a un linfoma progresivo. No se encontraron diferencias entre los grupos.
Bortezomib en el mieloma múltiple
En el mieloma múltiple (MM), el tratamiento de inducción con bortezomib más mantenimiento sigue mejorando la supervivencia de los pacientes sintomáticos recién diagnosticados. Este es el resultado del estudio hOVON65, cuyos resultados a largo plazo fueron presentados por el doctor Pieter Sonneveld, del Erasmus Medisch Centrum de Rotterdam. En este estudio, 827 pacientes con MM fueron aleatorizados a VAD (vincristina, doxorrubicina, dexametasona; n=414) o PAD (bortezomib, doxorrubicina, dexametasona; n=413) tras la terapia de inducción, seguida de dosis altas de melfalán y trasplante autólogo de células madre. La terapia de mantenimiento consistió en 50 mg diarios de talidomida (para el grupo VAD) o 1,3 mg de bortezomib/m2 iv quincenalmente (para el grupo PAD) durante dos años. La respuesta durante el tratamiento del protocolo pareció mejorar ligeramente ahora que todos los pacientes han completado el tratamiento: La remisión completa (RC) más la remisión completa nodular (RCn) se produjo en el 49% con DAP y en el 35% con DVA. Se observó una remisión parcial muy buena (RPMB) en el 26% y el 21%, y una remisión parcial ≥ (RP) en el 91% y el 83%, respectivamente. Tras un seguimiento medio de 67 meses, 111 de los pacientes tratados con DAV y 131 de los tratados con DAP estaban libres de progresión y vivos. La supervivencia libre de progresión (el tiempo transcurrido desde la aleatorización hasta la progresión, la recaída o la muerte) fue mejor con la PAD (ajustada al estadio) (ISS) (HR 0,78; IC 95% 0,66-0,91; p=0,002). La PAD también fue superior para el resultado secundario de la supervivencia global tras el ajuste de la ISS (HR 0,80; IC del 95%: 0,65-1,00; p=0,047).
Nueva mutación en ET JAK-2 negativa y PMF
Investigadores austriacos e italianos han descubierto una nueva mutación específica de los pacientes con trombocitopenia esencial (TE) JAK-2 negativa y mielofibrosis primaria (PMF). La alteración genética más común en el síndrome mieloproliferativo (MPN) es la mutación JAK2-V617F. Esto ocurre en el 95% de los pacientes con policitemia vera (PV) y en el 50-60% de los pacientes con ET y PMF. Las mutaciones en el exón 12 del JAK2 y en el gen del receptor de trombopoyetina MPL se observan en otro 5-10% de los casos. En los últimos años, se ha puesto de manifiesto que varios otros genes están afectados en la NMP. Sin embargo, estas mutaciones también se encuentran en otras enfermedades mieloides. Un marcador molecular específico para el 40% restante de pacientes con ET y PMF con JAK2 y MPL de tipo salvaje es, por tanto, muy bienvenido.
Thorsten Klampfl, del Centro de Investigación de Medicina Molecular de la Academia Austriaca de Ciencias de Viena, y sus colegas identificaron nuevas mutaciones en pacientes con PMF con JAK2 y MPL de tipo salvaje mediante la secuenciación del exoma completo. El análisis mostró inserciones y deleciones somáticas recurrentes en el CALR que codifica la calreticulina. Todas las mutaciones detectadas resultaron de un desplazamiento del marco de lectura y se agruparon en el último exón (exón 9) del gen. Tras este descubrimiento, los investigadores desarrollaron una prueba basada en la PCR que utilizaron para examinar a 1.107 pacientes con NMP en busca de mutaciones de inserción/deleción en el exón 9 de CALR. No se encontraron mutaciones en los pacientes PV, pero sí en los pacientes ET y PMF: las mutaciones CALR eran mutuamente excluyentes con JAK2 mutado y MPL mutado. Entre los pacientes con JAK2 y MPL de tipo salvaje, el 67% de los pacientes con ET y el 88% de los pacientes con PMF tenían CALR mutante. No se detectaron mutaciones del exón 9 de CALR en 254 pacientes con LMA de novo, en 45 con leucemia mieloide crónica, en 73 con síndrome mielodisplásico ni en 64 con leucemia mieloide crónica.
Los autores esperan que el marcador esté pronto a disposición de los clínicos para mejorar las decisiones diagnósticas y terapéuticas en la NMP.
Fuente: 55ª Reunión Anual de la ASH, 7-10 de diciembre de 2013, Nueva Orleans
InFo Oncología y Hematología 2014; 2(3): 30-32
Especial del Congreso 2014; 5(2): 6-7









