Los síndromes antisintetasa son un subgrupo de las miopatías inflamatorias idiopáticas y representan aquí alrededor del 35-40% de los casos. Sin embargo, si se detecta a tiempo, el pronóstico a largo plazo para los afectados es relativamente bueno. Un experto le explicó a qué síntomas debe prestar atención y cuáles son las opciones terapéuticas.
Los síndromes antisintetasa (TEA) son enfermedades raras con una prevalencia de 1,5-9/100.000 y una incidencia de 1,25 -2,5/1 millón. La edad media de aparición oscila entre 48 y 55 años, con un amplio rango en la primera manifestación (de 20 a 80 años). Las mujeres se ven afectadas con más frecuencia que los hombres.
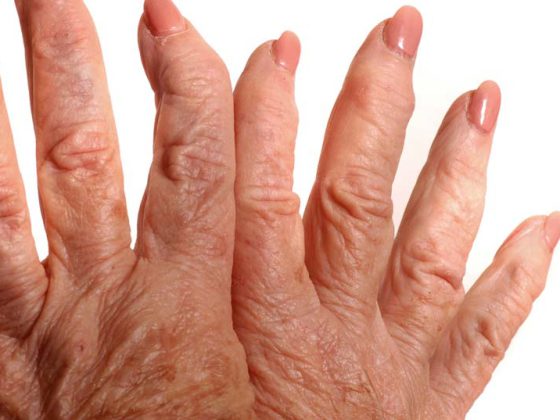
La enfermedad se define por la presencia serológica de un anticuerpo antisintetasa y clínicamente por la combinación de al menos uno de los tres síntomas principales miositis, enfermedad pulmonar intersticial o artritis, explicó la Dra. Jutta Bauhammer, Baden-Baden (D). Además, pueden aparecer síntomas secundarios como fiebre, síndrome de Raynaud o signos cutáneos como las llamadas manos de mecánico (Fig. 1) . Al principio de la enfermedad, a menudo sólo algunos de estos síntomas orgánicos están presentes al mismo tiempo. La tríada completa de miositis, afectación pulmonar y artritis es más frecuente en el síndrome de Jo-1, donde sólo está presente en el 20% de las primeras manifestaciones. En el curso posterior, alrededor del 50% se ven afectados, el seguimiento es máximo. 16 años.

Diagnósticos de laboratorio
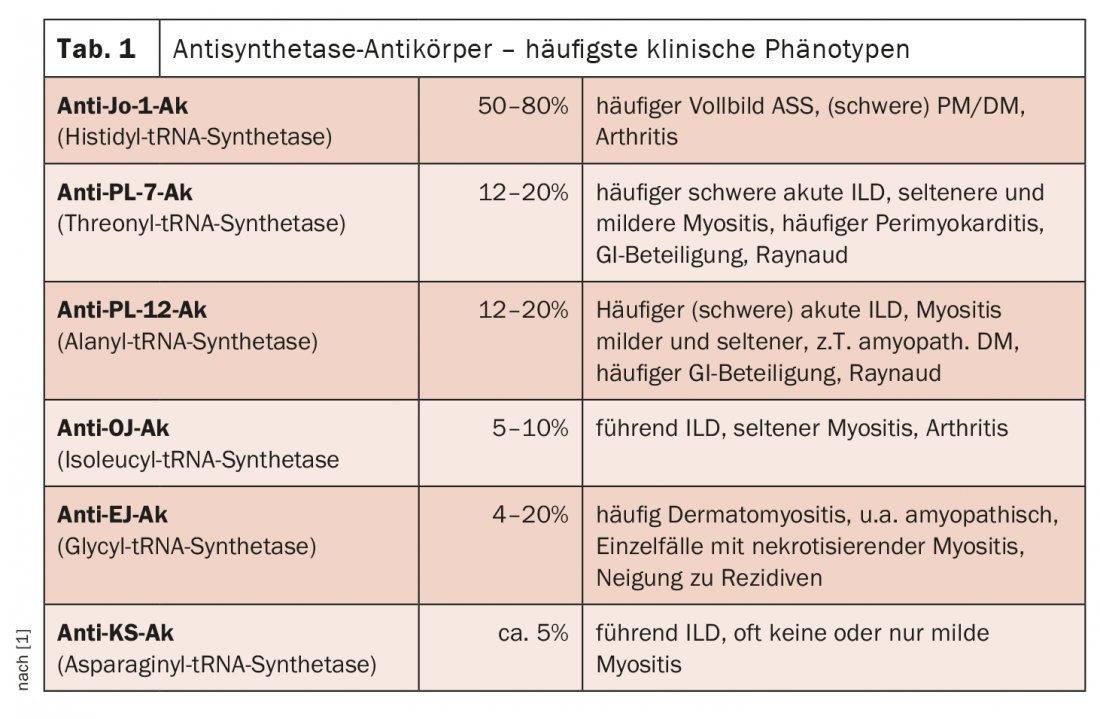
Desde el punto de vista químico-laboratorial, se conocen 11 anticuerpos antisintetasa (ASS-Ak), pero hasta ahora sólo 8 de ellos han sido asignados al cuadro clínico de un síndrome antisintetasa (Tab. 1). Los antígenos diana son las aminoacil-ARNt sintetasas, y dependiendo de cuál represente exactamente el antígeno, existen diferentes anticuerpos. Estas sintetasas son enzimas del citoplasma que intervienen en la traducción, catalizan la unión de un aminoácido a su ARNt específico. En general, sólo hay un ASS-Ak por paciente, son mutuamente excluyentes. La aparición simultánea de varios ASS-Ak se considera una rareza. El ASS-Ak más común en los pacientes caucásicos es el anticuerpo anti-Jo-1, presente en el 50-80% de los individuos afectados y dirigido contra la histidil-ARNt sintetasa. En este Ak, es más común ver un síndrome antisintetasa completo (es decir, los tres síntomas principales están presentes con afectación muscular, pulmonar y articular) con una miositis a menudo grave y en muchos casos una afectación articular más grave con artritis manifiesta.
Participaciones en órganos
La miositis está presente en el 75-80% de los casos de Jo-1-Ak, y en aproximadamente el 60% de los casos de otros anticuerpos (en los que el pulmón suele ser el órgano principal). Clínicamente, el cuadro suele ser de polimiositis, más raramente de dermatomiositis, y la miocarditis concomitante se observa en el 3-4% de los pacientes. La histología muestra típicamente una inflamación dominante del tejido conjuntivo perimisio con un infiltrado de abundantes macrófagos CD68 positivos como marcador (“miositis dominada por macrófagos”), puede producirse necrosis perifascicular, la densidad capilar suele ser anodina. También existen informes de casos aislados de miositis necrotizante.
La afectación articular se produce en el 70% de los Jo-1-Ak, y en torno al 50% de los no Jo-1-Ak. En el caso de los anticuerpos Jo-1, representa la primera manifestación orgánica aislada en el 20-35%. En la mayoría de los casos es poliarticular-simétrica (70%), también con afectación de las articulaciones pequeñas y medianas, y más raramente oligoarticular (30%). Algunos pacientes también presentan artralgias inflamatorias primarias. A diferencia de la artritis reumatoide (AR), estos pacientes presentan el síndrome de Raynaud con mucha más frecuencia (30-50%, los pacientes con AR aproximadamente el 10%). “Ahora se vuelve aún más insidioso”, advirtió el Dr. Bauhammer: en casos raros, los pacientes con síndrome antisintetasa también pueden tener anticuerpos anti-CCP positivos, que en realidad están calificados como muy específicos para la AR. En un estudio francés, este era el caso en un 6-9%. Cuando se examinó a los pacientes con artritis aislada, tenían serología positiva para la AR hasta en un 30% (ACPA en un 28%). Estos pacientes tienen una afectación articular del 100%, muy posiblemente con erosiones.
Los pulmones son uno de los órganos que determinan el pronóstico. La afectación pulmonar se produce en forma de enfermedad pulmonar intersticial (EPI), es decir, alveolitis o fibrosis, en el 60-80% con anticuerpos Jo-1, con mayor frecuencia (>80%) en pacientes sin Jo-1-Ak. Puede ser clínicamente aguda (10-30%) con inicio (per)agudo, progresión rápida con posible deterioro en pocos días hasta el punto de requerir cuidados intensivos o ventilación, fiebre frecuente y a veces insuficiencia respiratoria aguda. El 40-50% de los casos son crónicos, con un inicio insidioso y una progresión lenta hasta que se desarrolla una disnea de alivio, tos irritativa y reducción de la función pulmonar. El 20-25% de los pacientes son asintomáticos y sólo presentan anomalías en las lecturas o en los hallazgos del TAC. Si los afectados sobreviven a la fase aguda, el pronóstico a largo plazo es el mismo para las tres formas de progresión. No hay que olvidar que la hipertensión pulmonar (HP) también puede aparecer en los síndromes antisintetasa. Esto ocurre en casi el 10% de los casos con una Jo-1-Ak y en hasta el 20% de los pacientes con una no-Jo-1-Ak y también puede ser independiente de la propia enfermedad pulmonar intersticial, por ejemplo como HP “desproporcionada”, a veces también como HP primaria.
La latencia del diagnóstico de la HP en comparación con el diagnóstico inicial del síndrome antisintetasa es muy alta. En algunos casos, el diagnóstico sólo se realizó cuando la HP ya estaba muy avanzada. Según el Dr. Bauhammer, esto podría deberse principalmente a que los médicos piensan demasiado tarde en la posibilidad de una hipertensión pulmonar. En consecuencia, el pronóstico es malo cuando se produce HP. La tasa de supervivencia a 3 años aquí es sólo del 58%. En comparación con los pacientes sin HP, los que padecen hipertensión pulmonar presentan una disminución marcada de la DLco/FVC y es más probable que presenten Raynaud y microscopía capilar anormal.
Los signos cutáneos en los síndromes antisintetasa son las llamadas manos de mecánico o -cuando los signos aparecen en los pies- los “pies de excursionista” (Fig. 2 ). Aproximadamente un tercio de los pacientes están afectados. Se caracterizan por hiperqueratosis y eritema no pruriginoso, especialmente en la cara radial-palmar de los dedos y las palmas de las manos. A menudo existen asociaciones con enfermedades pulmonares intersticiales (graves). Las lesiones cutáneas remiten bajo terapia sistémica, casi nunca es necesaria una terapia local específica.
Los síntomas secundarios del ASA, es decir, Raynaud o fiebre, están presentes en el 30-40% de los pacientes cada uno. El síndrome de Raynaud no suele ser tan grave como la esclerodermia, pero existen informes de casos de úlceras digitales e incluso de isquemia digital aguda grave. La fiebre es más frecuente en los pacientes que presentan una afectación pulmonar aguda. El experto también informó sobre un estudio que evaluó una nueva aparición de Raynaud, manos de mecánico o fiebre en el curso de la enfermedad como presagio de una recaída aguda y/o una nueva manifestación orgánica (riesgo 4 veces mayor con Jo-1-Ak).
Terapia
La terapia depende en gran medida de la afectación del órgano, es decir, los inmunosupresores y los esteroides se seleccionan en función del órgano afectado y de su gravedad. En consecuencia, también existe una gama bastante amplia de medicamentos que pueden utilizarse. En la enfermedad pulmonar intersticial, que es un órgano importante desde el punto de vista del pronóstico, han demostrado ser muy eficaces, en particular, los inhibidores de la calcineurina y, en función de la gravedad, la ciclofosfamida o el rituximab. También hay informes de casos de buena respuesta al MMF o a las inmunoglobulinas intravenosas, aunque los datos sobre estas últimas son aún muy escasos en los casos de afectación pulmonar. Los inhibidores del TNF-alfa se han utilizado en la afectación articular, pero la miositis y la enfermedad pulmonar intersticial pueden empeorar con ellos (progresión en ≥33%). Desde 2020, existe en Alemania una nueva opción de utilizar nintedanib para la fibrosis pulmonar progresiva, que está aprobado para la esclerosis sistémica con afectación pulmonar desde marzo y para otras enfermedades pulmonares intersticiales crónicas fibrosantes y progresivas desde julio y, por tanto, potencialmente también para los pacientes con antisintetasa. Según los consejos del Dr. Bauhammer, siempre debe tenerse en cuenta una buena terapia de acompañamiento (gimnasia respiratoria, fisioterapia).
Previsión
El curso de la miositis suele considerarse más leve en comparación con, por ejemplo, el curso de la miositis con anticuerpos SRP. Los pacientes que presentan afectación pulmonar suelen tener una mayor mortalidad que los que no la presentan. Los individuos afectados con afectación pulmonar en un TEA -incluso si se desarrolla fibrosis- tienen una supervivencia significativamente mejor en comparación con los pacientes con fibrosis pulmonar idiopática, incluso con un patrón UIP en las imágenes o la histología.
Mensajes para llevarse a casa
- Recuerde que el paciente puede tener ASA, a menudo de inicio mono/oligosintomático.
- Indicativos de ASA en artritis aislada: síndrome de Raynaud, anti-Ro52-Ak y/o manos/pies de mecánico, entre otros.
- La nueva aparición de Raynaud, signos cutáneos, fiebre puede predecir una recaída o una nueva manifestación orgánica
- Elección de la inmunosupresión en función de la afectación orgánica, demostrada en la EPI: Inhibidores de la calcineurina, RIX
- No olvide el cribado del PH
- Previsión relativamente buena
– FomF Reuma Nefro Actualización (en línea), 29-31.10.2020
Fuente:
- Conferencia “Síndromes antisintetasa“ en el FomF Rheuma Nephro Refresher (en línea), 29.10.2020
InFo PAIN & GERIATURE 2020; 2(2): 42-43 (publicado el 10.12.20, antes de impresión).