Los antihistamínicos H1 se dividen en una primera generación más antigua con efectos secundarios sedantes y una 2ª generación sin estos efectos. Nuestro autor arroja luz sobre la farmacología de este grupo de sustancias activas.
Hasta ahora se conocen cuatro receptores de histamina diferentes. Estas moléculas acopladas a la proteína G se localizan en la superficie celular y ejercen diferentes efectos en función del lugar de expresión. Mientras que la activación del receptor H1 provoca en particular prurito, vasodilatación, contracción de la musculatura lisa con broncoespasmo o calambres abdominales, secreción de moco con rinorrea y aumento de la secreción bronquial, así como un aumento de la permeabilidad vascular, los receptores H2 están implicados en particular en el aumento de la secreción de jugo gástrico y ácido. Además, existen receptores H3, que desempeñan un papel en el SNC como autorreceptores presinápticos, y receptores H4, que intervienen en la diferenciación y modulación de las células inmunitarias, entre otras cosas. En este artículo se abordan las sustancias dirigidas contra el receptor H1, mientras que no se tratan los antagonistas del receptor H2, de los que sólo la ranitidina sigue comercializándose en Suiza. Se están desarrollando clínicamente agonistas y antagonistas de los receptores H3 y H4.
Farmacodinámica de los antihistamínicos H1
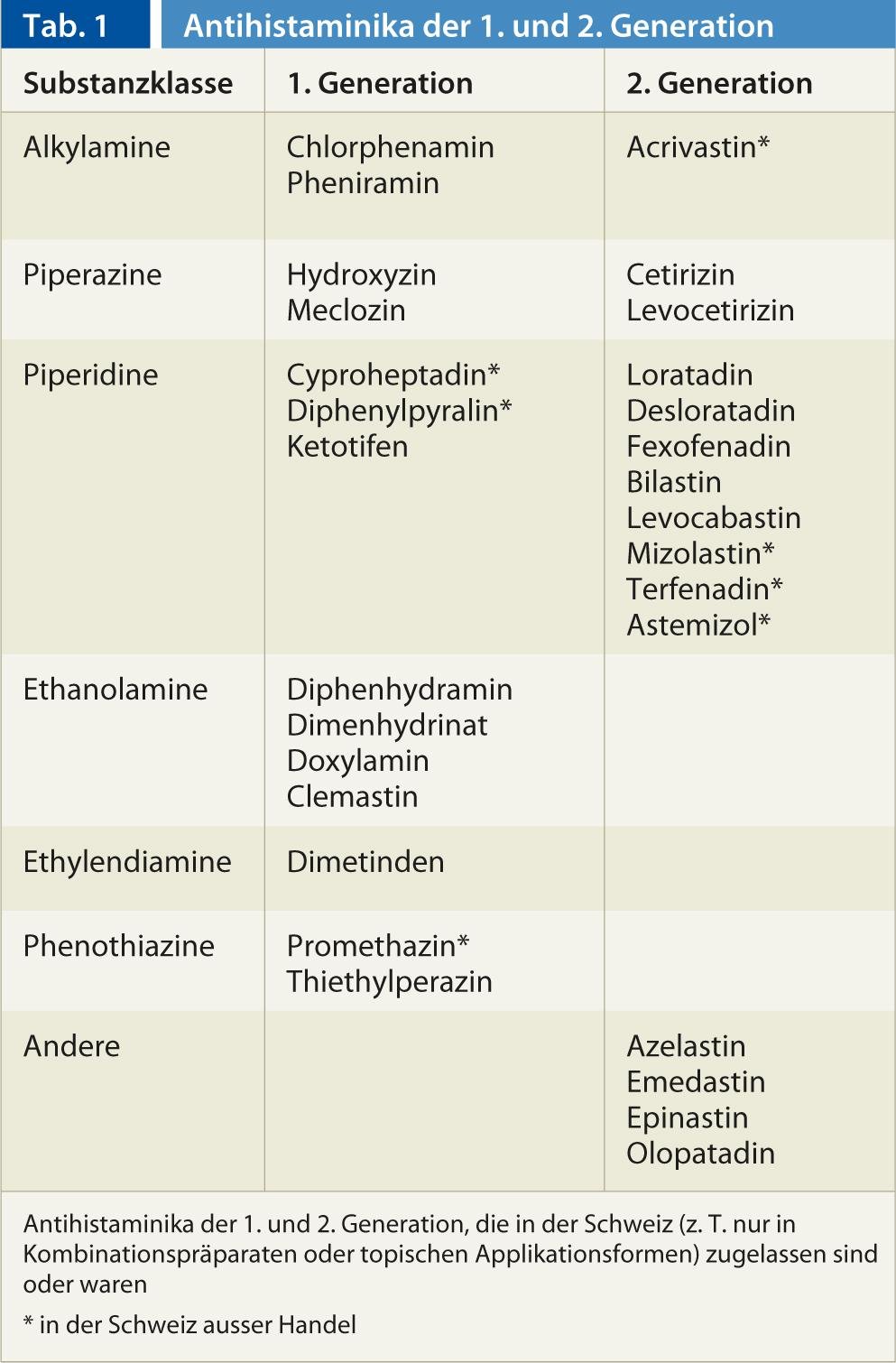
En Suiza se dispone de 22 sustancias activas de la clase de los antihistamínicos H1. En la actualidad, se supone que los antihistamínicos estabilizan el receptor H1 en su conformación inactiva y garantizan así que la histamina pueda activar menos receptores. Mientras que los antihistamínicos H1 de la primera generación, la más antigua, penetran bien en el sistema nervioso central y ejercen allí un efecto sedante en los receptores H1 postsinápticos, no ocurre lo mismo con los representantes de la segunda generación o no en concentraciones terapéuticas (Tab. 1 ). Debido a su buena eficacia en los receptores H1 centrales, algunos representantes del Sedantes/hipnóticos de 1ª generación (por ejemplo, doxilamina, difenhidramina), antieméticos (por ejemplo, meclozina) o contra el mareo (por ejemplo, dimenhidrinato). La escasa penetración en el SNC de los representantes del La segunda generación se debe al hecho de que estas sustancias son hidrófilas y sustratos del transportador dirigido hacia el exterior P-glicoproteína presente en la barrera hematoencefálica (entre otras barreras de membrana del organismo). Esto evita la sedación, que se utiliza en indicaciones antialérgicas para sustancias del La primera generación a menudo limitaba la terapia. Algunos antihistamínicos H1 del 1ª generación tienen efectos adicionales sobre los receptores de acetilcolina, noradrenalina y serotonina, mientras que los representantes de la La 2ª generación inactiva específicamente el receptor H1.
En general, la eficacia clínica de los antihistamínicos H1 del 1. generación han sido poco estudiados en ensayos clínicos, mientras que las pruebas del uso de antihistamínicos H1 de la La 2ª generación es buena para la rinitis alérgica, la conjuntivitis alérgica y la urticaria. El uso de productos de la La 2ª generación en dermatitis atópica, asma, anafilaxia, angioedema no alérgico, resfriados, prurito de origen no alérgico, etc. ha sido poco investigada en estudios o éstos no mostraron efectos convincentes y tampoco existe aprobación para tales indicaciones. En los niños, las sustancias del La primera generación puede provocar efectos secundarios a veces amenazadores, por lo que la indicación debe hacerse con especial cuidado. En la llamada 3ª generación, los enantiómeros o metabolitos de moléculas del 2ª generación, sin que existan diferencias farmacodinámicas importantes, por lo que las sustancias son farmacológicamente similares a las 2ª generación.
Farmacocinética de los antihistamínicos H1
El primer obstáculo que debe superar un medicamento para ser eficaz es su toma. Garantizar el cumplimiento o la adherencia es, por tanto, de especial importancia en la práctica médica diaria. La mayoría de los antihistamínicos H1 de uso sistémico están disponibles como formas farmacéuticas sólidas (comprimidos, grageas, comprimidos recubiertos con película, supositorios) o gotas para administración oral. Algunas sustancias se aplican localmente (por ejemplo, colirios). Sólo unos pocos pueden administrarse también por vía intravenosa (dimetinden, clemastina, tietilperazina). A la reabsorción, que es rápida para la mayoría de los antihistamínicos H1 de 2ª generación y conduce a niveles máximos al cabo de una a tres horas, le sigue la distribución en la sangre y los tejidos, el metabolismo, si es necesario, y la excreción.
Existen grandes diferencias entre los antihistamínicos, especialmente en lo que respecta a su metabolización y eliminación (Tab. 2). Además, existen diferencias en el metabolismo de las personas causadas por influencias genéticas y medioambientales. En particular, la enzima CYP2D6 del citocromo P450, implicada en la degradación de algunos antihistamínicos H1 (Tab. 2) , muestra una fuerte variabilidad genética, que puede ir desde la ausencia hasta la multiplicación de la actividad enzimática normal. Además, el CYP2D6 puede ser inhibido por algunas sustancias: el bupropión, el cinacalcet, los inhibidores selectivos de la recaptación de serotonina paroxetina, fluoxetina y duloxetina, así como el fármaco antifúngico terbinafina se encuentran entre los inhibidores más potentes del CYP2D6. Sin embargo, dado que todos los antihistamínicos H1 son degradados por más de una enzima y a menudo se eliminan por vía renal sin cambios, la variabilidad genética y la inhibición del CYP2D6 muy raramente desempeñan un papel en el uso de los antihistamínicos H1.
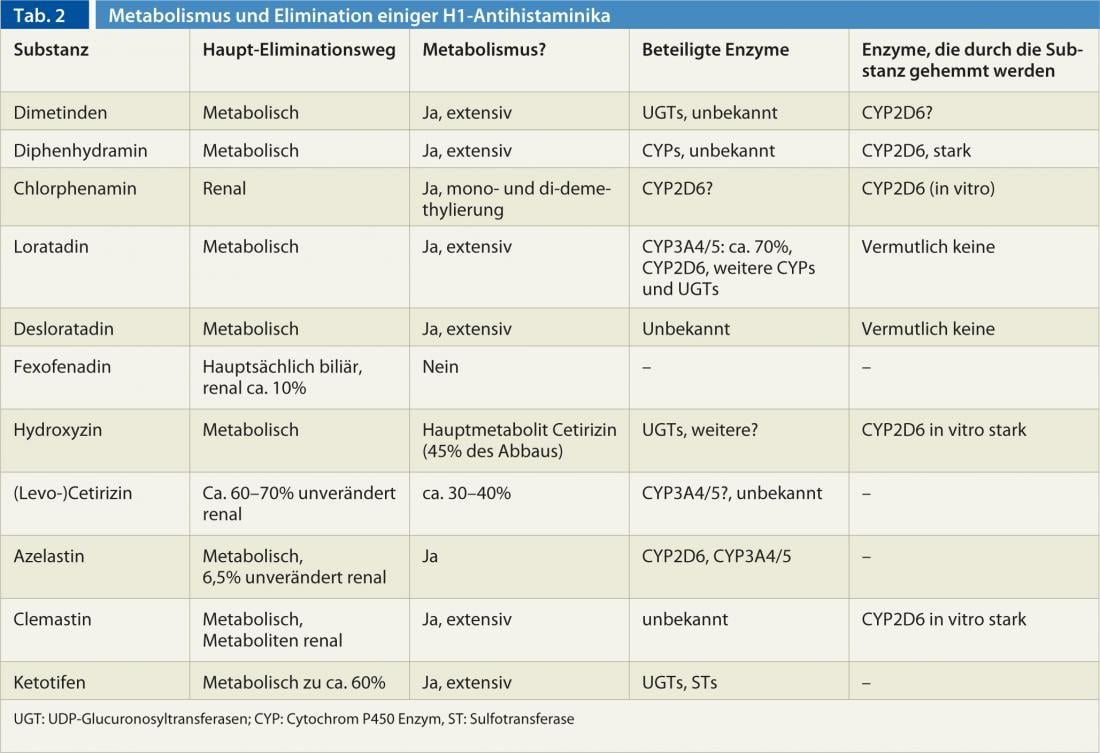
Algunos antihistamínicos H1 de 1ª generación son también inhibidores del CYP2D6. (Tab. 2), por lo que cuando se utilizan concomitantemente sustratos del CYP2D6 (por ejemplo, codeína, dextrometorfano, muchos antipsicóticos [haloperidol, risperidona, aripiprazol], atomoxetina, muchos antidepresivos [la mayoría de los tricíclicos, venlafaxina, etc.], así como los betabloqueantes metoprolol, carvedilol y timolol), debe elegirse una dosis más baja del sustrato para evitar efectos adversos.
En general, la farmacocinética de las sustancias de la 1ª generación sólo se conoce de forma incompleta. sólo se conoce de forma incompleta, ya que las sustancias se aprobaron hace décadas con muchos menos datos de los necesarios hoy en día. Pero también existen lagunas en los conocimientos sobre los preparados de 2ª generación: La desloratadina se metaboliza en el metabolito igualmente activo 3-hidroxi-desloratadina, pero se desconoce la enzima o enzimas implicadas, aunque se ha descubierto que el 2% de los europeos de y hasta el 20% de los africanos no pueden formar 3-hidroxi-desloratadina. Las enzimas CYP2D6 y CYP3A4/5 implicadas en el metabolismo de la loratadina no parecen ser responsables de ello.
La CYP3A4 es la enzima citocromo P450 más importante, tanto en cantidad como en número de sustratos. Aunque no se conocen variantes genéticas que alteren la función del CYP3A4 (a pesar de las intensas investigaciones), existen fármacos que aumentan la actividad enzimática (rifampicina, efavirenz, fenitoína, carbamazepina, ingredientes de la hierba de San Juan, etc.) y la inhiben (antifúngicos azólicos, eritromicina, claritromicina, ritonavir, verapamilo, diltiazem, amiodarona, etc.).(antifúngicos azoles, eritromicina, claritromicina, ritonavir, verapamilo, diltiazem, amiodarona, ingredientes del pomelo, especialmente en el zumo de pomelo, etc.). En cambio, el CYP3A5, que descompone esencialmente los mismos fármacos que el CYP3A4, casi nunca está presente en los europeos debido a una variante genética, mientras que suele ser funcional en los africanos. A excepción de la loratadina, la azelastina y probablemente también la cetirizina, el CYP3A4/5 no interviene en los antihistamínicos.
Fue aún más sorprendente cuando se demostró un efecto del zumo de pomelo en un estudio con fexofenadina: Cuando la fexofenadina se tomó junto con zumo de pomelo, los niveles de fexofenadina disminuyeron, especialmente poco después de tomarla, en comparación con la toma con agua (se habría esperado un aumento si el CYP3A4 fuera inhibido por las furanocumarinas del zumo de pomelo). Este efecto puede explicarse por el hecho de que otras sustancias del zumo de pomelo (el flavonoide naringina) inhiben el transportador intestinal de entrada OATP1A2, necesario para la absorción de la fexofenadina desde la luz intestinal.
Otra interacción que aún no se ha aclarado del todo también afecta a la fexofenadina: la administración simultánea de itraconazol dio lugar a niveles de fexofenadina mucho más elevados . Dado que la fexofenadina no es metabolizada por el CYP3A, que es potentemente inhibido por el itraconazol, la interacción se explicó por la inhibición de la glicoproteína P, el transportador que se cree que desempeña un papel importante en la eliminación de la fexofenadina. Aún debe investigarse si esta hipótesis es correcta y otros antifúngicos azólicos también provocan aumentos de nivel.
Por otro lado, debe tenerse en cuenta que sustancias como la (levo-)cetirizina se eliminan principalmente por vía renal, por lo que las restricciones de la función renal también deben conllevar reducciones de la dosis. Para la levocetirizina, por ejemplo, es necesario reducir la dosis a partir de un tasa de filtración glomerular inferior a 50 ml/min, se prescribe una administración de 5 mg cada 2 días; en caso de deterioro más grave de la función renal, el intervalo de dosificación debe prolongarse aún más.
En Suiza, la información relativa a los pacientes sobre efectos secundarios e interacciones, ajustes de dosis y otros problemas relacionados con los medicamentos puede obtenerse en los servicios de farmacología clínica de los hospitales universitarios.
Efectos adversos de los antihistamínicos: Enfoque del tiempo QTc
Mientras que la mayoría de los efectos adversos de los antihistamínicos H1 de se deben a su acción en el receptor H1 (fatiga, disminución del rendimiento cognitivo y psicomotor, aumento del apetito) o (en el caso de las sustancias más antiguas) por efectos en el receptor de la m-acetilcolina (sequedad de boca, retención urinaria, taquicardia), en el alfa-adrenoceptor (hipotensión, mareo, taquicardia refleja) o en el receptor de la serotonina (por ejemplo, aumento del apetito). Mientras que los efectos de los antihistamínicos H1 pueden explicarse por sus efectos sobre el receptor alfa-adrenérgico (hipotensión, mareos, latidos reflejos) o sobre el receptor de la serotonina (por ejemplo, aumento del apetito), es menos conocido que algunos antihistamínicos H1 también inhiben los canales iónicos cardíacos, especialmente el canal IKr, que modula la salida rápida de potasio durante la repolarización y puede provocar así una prolongación de la repolarización (del intervalo QT en el ECG) e incluso taquicardias ventriculares torsades de pointes.
En general, el potencial de los antihistamínicos H1 para prolongar el tiempo QTc en el ECG está poco estudiado. El interés de por este efecto secundario potencialmente mortal se despertó cuando se recibieron varios informes de episodios de torsades de pointes en por los primeros antihistamínicos H1 de 2ª generación, la terfenadina y el astemizol. En la mayoría de los casos de , se habían tomado sobredosis o no se habían observado interacciones que aumentaran la concentración. Las dos sustancias fueron retiradas del mercado en 1990 debido a las arritmias ventriculares. Para la loratadina, fexofenadina y cetirizina, existen informes de casos individuales de prolongación del tiempo QTc y en algunos casos taquicardia torsades de pointes.
Los factores de riesgo generales para la prolongación del tiempo QTc se muestran en la Tabla 3 . Por lo tanto, parece aconsejable, sobre todo en los pacientes de riesgo a los que se administran antihistamínicos con regularidad y a dosis elevadas, realizar un ECG y controlar el potasio y el magnesio séricos.

CONCLUSIÓN PARA LA PRÁCTICA
- Los antihistamínicos H1 de 2ª generación suelen tolerarse bien.
- La prolongación del tiempo QTc es posible, especialmente con sobredosis y factores de riesgo.
- El zumo de pomelo no debe tomarse con fexofenadina, ya que reduce las concentraciones de fexofenadina. El itraconazol aumenta las concentraciones de fexofenadina, lo que puede provocar síntomas de sobredosis.
- En el caso de los antihistamínicos de excreción principalmente renal, como la (levo-)cetirizina, la dosis debe ajustarse en caso de insuficiencia renal.
PD Alexander Jetter, MD
Literatura:
- Simons FE, Simons KJ: Histamina y antihistamínicos H1: celebrando un siglo de progreso. J Allergy Clin Immunol 2011; 128: 1139-1150.
- Shon JH, Yoon YR, Hong WS, Nguyen PM, Lee SS, Choi YG, Cha IJ, Shin JG: Efecto del itraconazol sobre la farmacocinética y la farmacodinámica de la fexofenadina en relación con el polimorfismo genético MDR1. Clin Pharmacol Ther 2005; 78: 191-201.
- Banfield C, Gupta S, Marino M, Lim J, Affrime M: El zumo de pomelo reduce la biodisponibilidad oral de la fexofenadina pero no de la desloratadina. Clin Pharmacokinet 2002; 41: 311-318.
- Hondeghem LM, Dujardin K, Hoffmann P, Dumotier B, De Clerck F: La prolongación del QTc inducida por fármacos subestima peligrosamente el potencial proarrítmico: lecciones de la terfenadina. J Cardiovasc Pharmacol 2011; 57: 589-597.
- Compalati E, Baena-Cagnani R, Penagos M, Badellino H, Braido F, Gómez RM, Canonica GW, Baena-Cagnani CE: Revisión sistemática sobre la eficacia de la fexofenadina en la rinitis alérgica estacional: un metaanálisis de ensayos clínicos aleatorizados, doble ciego y controlados con placebo. Int Arch Allergy Immunol 2011; 156: 1-15.
PRÁCTICA GP 2013; 8(3): 14-17