La hemoglobinuria paroxística nocturna (HPN) es una enfermedad rara adquirida que se caracteriza por hemólisis intravascular y hemoglobinuria. Debido a una pérdida prolongada de hemoglobina a través de la orina, la HPN puede provocar una carencia de hierro y aumentar la sospecha de anemia. Sin embargo, la HPN es una enfermedad extremadamente heterogénea con diversas manifestaciones y a menudo se solapa con otras enfermedades de la médula ósea.
La HPN es una enfermedad muy rara con una incidencia de aproximadamente 1,3/1.000.000 y una prevalencia de unos 16 por millón de habitantes. Los síntomas fueron descritos por primera vez como hemoglobinuria paroxística en 1882 por el Dr. Strübing de la policlínica médica de Greifswald. Marchiafava y Micheli introdujeron el término síndrome de Marchiafava-Micheli, que aún hoy se utiliza como sinónimo de la enfermedad. El nombre de HPN se acuñó por las tres características de esta enfermedad: Paroxística= se produce en episodios; Nocturna= se produce por la noche; Hemoglobinuria= hemoglobina en la orina. Hoy en día sabemos que la hemólisis también puede producirse sin síntomas y ser constante a lo largo del día. Además, sólo uno de cada cuatro pacientes presenta hemoglobina en la orina.
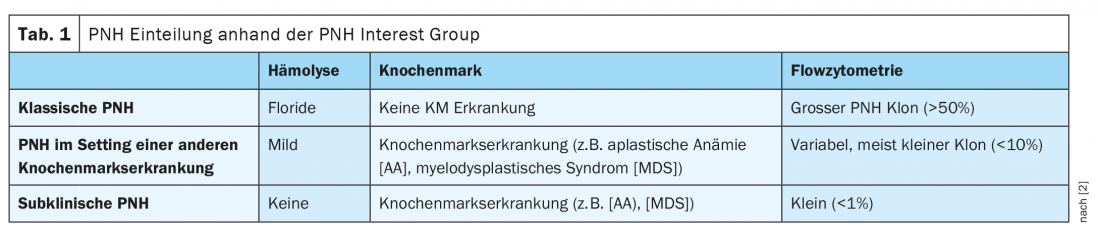
Puede dividirse en tres formas
La HPN puede dividirse en tres formas según el grado de hemólisis, los hallazgos en la médula ósea y la citometría de flujo. En la HPN clásica, el paciente presenta una hemólisis muy marcada pero ninguna otra enfermedad de la médula ósea. Además, estos pacientes tienen un gran clon de HPN >50%. Además de la HPN clásica, la enfermedad también puede darse en el contexto de otras enfermedades de la médula ósea, clásicamente en anemia aplásica. Estos pacientes no suelen ser sintomáticos y sólo presentan una hemólisis leve y un pequeño clon variable de HPN <10%. La HPN subclínica suele aparecer en enfermedades de la médula ósea, como la anemia aplásica o el síndrome mielodisplásico. Por lo general, en estos casos sólo se detecta un clon de HPN muy pequeño <1%, sin signos de hemólisis (Tab. 1) [2]. Además, la HPN puede solaparse con otras enfermedades en cualquier momento. Es posible una transición a la anemia aplásica (AA), así como a la leucemia mieloide aguda (LMA) y al síndrome mielodisplásico (SMD).
Siempre empieza con PIGA
La HPN se debe a una mutación ligada al cromosoma X del gen del fosfatidil inositol glicano A (PIGA). Esta enzima está implicada en la biosíntesis del llamado anclaje GPI. El anclaje GPI fija ciertas proteínas a la superficie de la membrana celular. Estas proteínas unidas incluyen en particular el factor acelerador de la descomposición del complemento (CD55) y el inhibidor de membrana de la lisis reactiva (CD59). Estos dos componentes sirven para defenderse del ataque complementario a la membrana eritrocitaria. La ausencia parcial o completa del anclaje GPI y la correspondiente omisión de las proteínas de superficie reguladoras del complemento significa que la membrana eritrocitaria está inadecuadamente protegida. El resultado es la hemólisis intravascular. Entre otras cosas, la hemólisis intravascular provoca la formación de hemoglobina libre en la sangre, que se une al óxido nítrico. El resultado es una disfunción endotelial con la consecuente contracción del músculo liso y la activación y agregación plaquetaria, lo que conlleva un mayor riesgo de tromboembolismo.
Cabe mencionar que los pacientes con una mutación PIGA suelen presentar también otras mutaciones somáticas. En hematología, hablamos entonces de cambios clonales o clones. Esta es una de las razones de los diferentes cursos, y en algunos casos puede desarrollarse una enfermedad maligna.
Triásico clásico
Los pacientes con HPN presentan una tríada clásica consistente en hemólisis, tromboembolismo y posible fallo de la médula ósea. Esto significa que las distintas quejas pueden producirse en momentos diferentes. Entre ellas figuran disfagia (41%), hipertensión pulmonar (47%), disnea (66%), dolor abdominal (59%), insuficiencia renal (64%), disfunción eréctil (47%), hemoglobinuria (26%) y trombosis (40%). Sin embargo, los principales síntomas son la anemia (88%) y la fatiga pronunciada (97%). Sin embargo, además de estos síntomas, también pueden producirse presentaciones atípicas, como ha demostrado un informe de un caso de Suiza. En este caso concreto, la HPN se detectó finalmente en un paciente con infarto de miocardio, lo que indicaba una trombosis arterial [3]. Otra publicación del New England Journal, informa sobre un paciente joven con cefalea, dolor abdominal, anemia y trombocitopenia, que tuvo una trombosis de la vena sinusal como complicación de la HPN [4]. Según la Dra. med. Beatrice Drexler, del Hospital Universitario de Basilea, las trombosis suelen ser muy relevantes en los pacientes con HPN porque influyen en la mortalidad. Suelen afectar no sólo a las venas profundas de las piernas, sino que pueden aparecer en localizaciones bastante atípicas [1].
Citometría de flujo como patrón oro
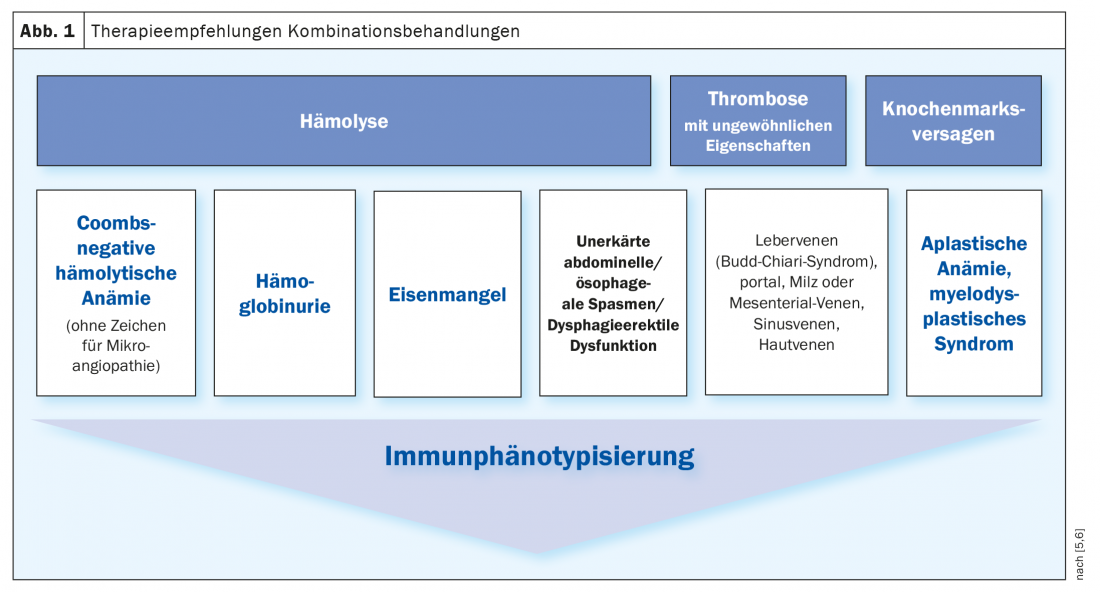
El patrón oro actual para la confirmación diagnóstica es la citometría de flujo a partir de sangre periférica, que detecta la falta de expresión de moléculas ancladas a GPI en eritrocitos y leucocitos. En este método, las células a examinar fluyen una tras otra a través de una delgada cámara de medición, la llamada célula de flujo. Determinados antígenos de la superficie de las células se marcan con anticuerpos monoclonales marcados con fluorescencia para poder medir la fluorescencia de cada partícula excitada por la luz láser. El criterio mínimo de diagnóstico es una población deficiente en GPI para al menos dos proteínas ancladas a GPI diferentes en dos líneas celulares distintas. También son relevantes el tipo y el tamaño del clon de HPN, que también pueden definirse en la citometría de flujo y proporcionar información sobre el riesgo de trombosis. Las indicaciones para la citometría de flujo son la hemólisis, la trombosis y la insuficiencia de la médula ósea (Fig. 1) [5,6].
Algoritmo terapéutico
En pacientes asintomáticos con valores de laboratorio discretos, no suele ser necesario ningún tratamiento. Sin embargo, estos pacientes deben ser objeto de un seguimiento regular mediante hemogramas, parámetros de hemólisis y citometría de flujo. Para los pacientes que necesitan tratamiento, la atención se centra en la terapia sintomática. La sustitución por ácido fólico es obligatoria, así como la administración de vitamina B12 y hierro, en función de los valores de laboratorio. Dependiendo del tamaño del clon y según sea necesario, pueden tomarse otras medidas de apoyo. Además, la inhibición de la vía terminal del complemento se realiza mediante un anticuerpo monoclonal como el eculizumab o el ravalizumab. Debe administrarse una infusión con estos inhibidores del complemento cada dos u ocho semanas. Esto puede inhibir la hemólisis intravascular mediada por el complemento, reducir la necesidad de transfusiones, mejorar la calidad de vida de los pacientes y también reducir el riesgo de episodios tromboembólicos, que son la principal causa de muerte en la HPN.
Mensajes para llevarse a casa
- La HPN es una enfermedad heterogénea con diversas manifestaciones.
- A menudo se solapa con otras enfermedades de la médula ósea (anemia aplásica y síndrome mielodisplásico).
- El patrón oro para el diagnóstico es la citometría de flujo a partir de sangre periférica.
- El pronóstico ha mejorado significativamente con la administración de inhibidores del complemento.
- Sin embargo, la terapia es muy costosa.
- Otras terapias modificadoras de la enfermedad están en desarrollo.
Literatura:
- Dra. med. Beatrice Drexler, Hospital Universitario de Basilea, “Was wenn Eisen nicht wegen Eisen mangelt?”, conferencia Iron Academy, 17.06.2021.
- Parker, et al: Diagnóstico y tratamiento de la hemoglobinuria paroxística nocturna . Sangre 2005, doi: 10.1182/blood-2005-04-1717.
- Gerber, et al: ¿Inhibición del complemento para tratar el infarto de miocardio? BMJ Case Rep. 2011, doi: 10.1136/bcr.01.2011.3701.
- Sykes, et al: Caso 40-2017: Una mujer de 32 años con cefalea, dolor abdominal, anemia y trombocitopenia. N Engl J Med. 2017, doi: 10.1056/NEJMcpc1710566.
- Röth, et al: Algoritmo clínico de cribado y diagnóstico de la hemoglobinuria nocturna: consenso de expertos. Eur J Haematol 2018, doi: 10.1111/ejh.13059.
- Borowitz, et al: Citometría B Clin Cytom 78:211-230. Drexler B Pipette – Swiss Laboratory Medicine 2010, nº 1/2 Marzo 2021.
PRÁCTICA GP 2021; 16(9): 20-22