Un tratamiento precoz y adecuado puede tener un efecto positivo en la evolución posterior de esta rara enfermedad multisistémica. Este artículo ofrece un breve resumen de lo que se sabe hoy en día. Entre otras cosas, la identificación de nuevos biomarcadores podría servir de base para una terapia personalizada en el futuro.
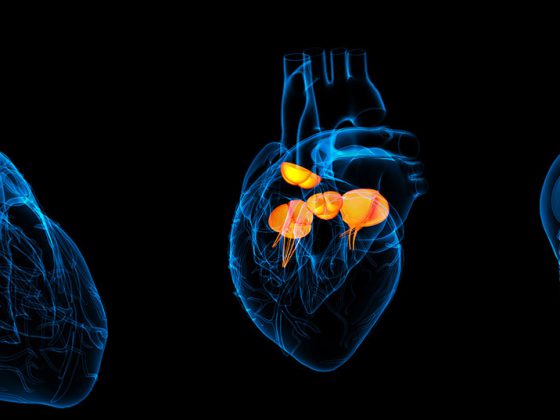
La dermatomiositis juvenil (DMJ) es una enfermedad sistémica e inmunomediada de la infancia caracterizada por debilidad muscular y erupciones cutáneas típicas [9]. Otros sistemas orgánicos, como el corazón y los pulmones, también pueden verse afectados y siguen siendo la causa más común de muerte en estos pacientes. Es la miopatía inflamatoria idiopática más frecuente en la infancia, con una incidencia de 2-4/millón/año.
El proceso inflamatorio se caracteriza por la infiltración del tejido afectado por células del sistema inmunitario, como las células T y las células dendríticas plasmocitoides, y una firma de interferón [1]. La mayoría de los síntomas se deben a la vasculopatía, que provoca la falta de funcionamiento y la pérdida de células endoteliales. Una inmunosupresión eficaz, normalmente una terapia combinada de metotrexato subcutáneo y corticoesteroides orales, ha reducido enormemente la mortalidad (actualmente en torno al 1%) y cerca del 50% de los pacientes sólo presentan un brote de la enfermedad antes de alcanzar la remisión [2]. El otro 50% presenta un curso crónico, con recaídas frecuentes, mayor morbilidad y menor calidad de vida [2].

Subfenotipado mediante biomarcadores
La DMJ es una enfermedad muy heterogénea con diferentes fenotipos, por lo que diversos sistemas orgánicos pueden verse afectados en distintos grados. Los autoanticuerpos han sido objeto de una intensa investigación en los últimos años y han permitido caracterizar mejor los diversos fenotipos; por lo tanto, los autoanticuerpos tienen valor pronóstico [3]. El seguimiento de la actividad de la enfermedad y del daño orgánico causado por la inflamación es una parte importante del tratamiento de estos jóvenes pacientes, ya que determina el resultado a largo plazo de esta enfermedad [4]. El clínico dispone de diversas herramientas para ello, como la histopatología del tejido muscular y cutáneo, la microscopía capilar (Fig. 1), las puntuaciones clínicas para evaluar la afectación muscular y cutánea [5], y ahora también diversos biomarcadores en la sangre [6,7]). Las puntuaciones clínicas utilizadas para evaluar la actividad de la enfermedad requieren la cooperación del paciente, lo que a veces supone un reto en los niños más pequeños. Estas puntuaciones clínicas también tienen una utilidad limitada para detectar la inflamación subyacente o para diferenciar otras causas de debilidad muscular, como la toxicidad de los esteroides.

Directrices para optimizar la gestión de la enfermedad
En 2012 se puso en marcha la iniciativa europea SHARE (Single Hub and Access point for paediatric Rheumatology in Europe) con el objetivo de estandarizar la gestión diagnóstica y terapéutica de los niños y adolescentes con enfermedades reumáticas en toda Europa. Para JDM, estas recomendaciones se publicaron en 2017 [6].
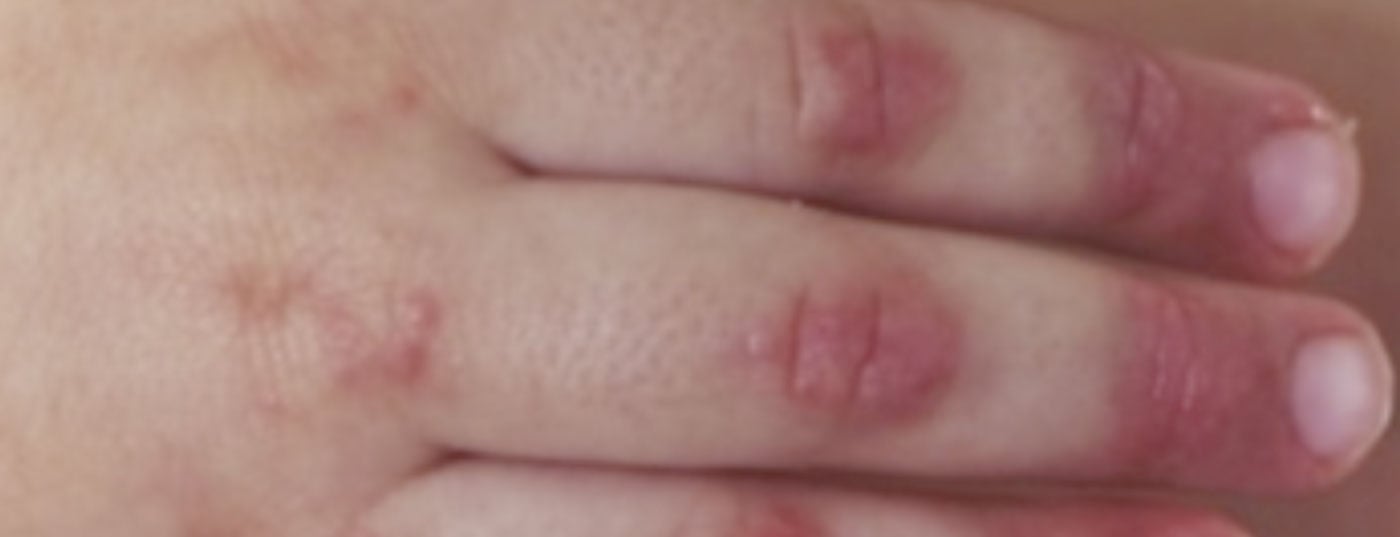
Diagnóstico: El diagnóstico de la dermatomiositis juvenil sigue siendo fundamentalmente clínico, con hallazgos cutáneos típicos (fig. 2) y debilidad muscular acentuada proximalmente. Por lo tanto, si se sospecha de DMJ, debe realizarse una derivación rápida a un centro de excelencia cercano. Una terapia temprana y adecuada puede prevenir daños a largo plazo y aumentar las posibilidades de una remisión precoz. Los criterios diagnósticos clásicos de Bohan y Peter de 1975 contienen cinco elementos:
- signos cutáneos característicos
- Debilidad muscular proximal
- biopsia muscular patológica típica
- enzimas musculares elevadas
- cambios miopáticos en el electromiograma (EMG)
→ En la práctica de la reumatología pediátrica, la EMG ha sido sustituida por la IRM no dolorosa (fig. 3) [8].
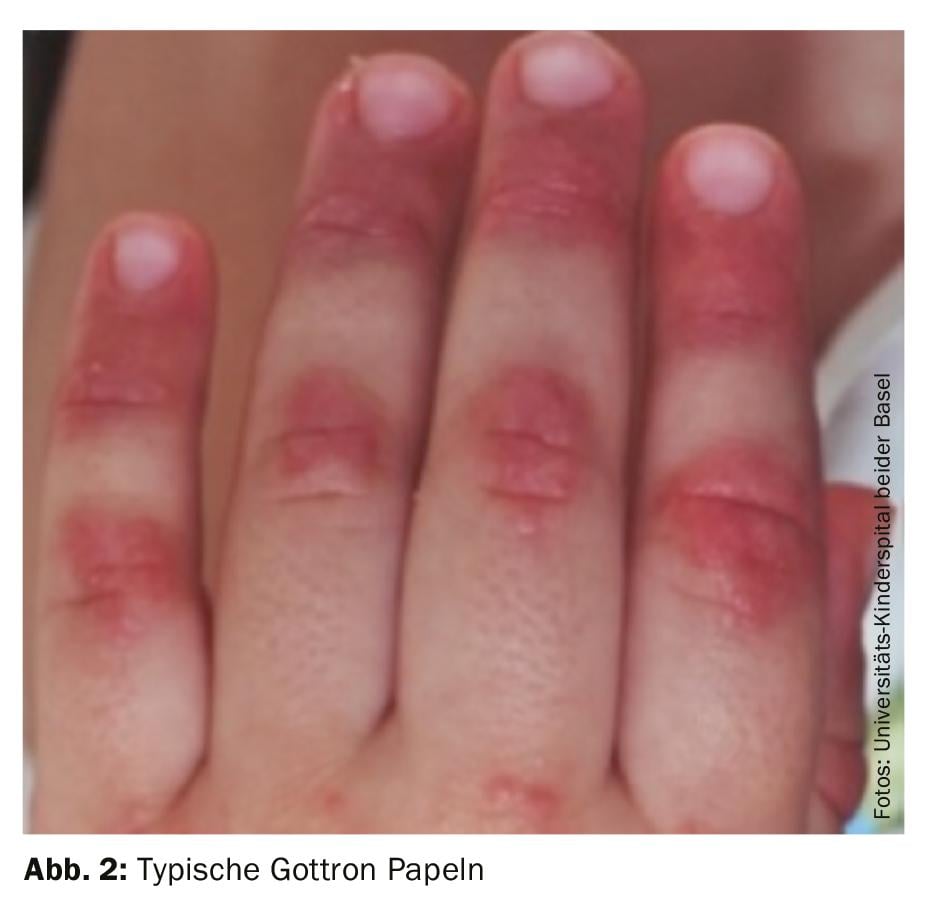

En 2017 se publicaron nuevos criterios de clasificación; por ejemplo, omiten la biopsia muscular para los síntomas cutáneos patognomónicos y también incluyen los autoanticuerpos (anti-Jo-1) en los criterios [9]. La ecografía también se utiliza cada vez más, ya que no requiere sedación en los niños más pequeños.
La debilidad muscular debe evaluarse en el niño con diferentes instrumentos. Existen pruebas musculares funcionales, las llamadas Children’s Myositis Activity Score (CMAS) [10], así como pruebas musculares que miden únicamente la fuerza, las llamadas Manual Muscle Test (MMT) [11]. Ambas pruebas son útiles para evaluar la debilidad muscular en el momento del diagnóstico; pero también sirven como importantes parámetros de seguimiento que pueden dar una indicación significativa de la actividad de la enfermedad. Ambas pruebas forman parte de los criterios de remisión de 2013 [12].
Wienke et al. han validado recientemente varios biomarcadores (galectina-9 y CXCL10) que desempeñan un papel no sólo en el diagnóstico, sino también en el seguimiento y pueden facilitar las decisiones de tratamiento al inicio de la enfermedad: En este estudio prospectivo, los pacientes con niveles elevados de estos biomarcadores en el momento del diagnóstico mostraron una evolución más grave en el seguimiento y, por tanto, podrían beneficiarse potencialmente de una terapia más intensiva. Por el contrario, en los pacientes con una sola recaída, los valores casi se habían normalizado ya unos meses después del diagnóstico bajo terapia. Además, se pudo demostrar que los marcadores ya mostraban un nuevo aumento en la sangre antes de los hallazgos clínicos de un brote de la enfermedad [7].

Terapia 2: La terapia de primera línea sigue siendo una combinación de esteroides y metotrexato, posiblemente ciclosporina [6]. El metotrexato debe mantenerse durante al menos 2 años; los esteroides se interrumpen en el primer año de terapia en el caso de un tratamiento monofásico [6]. Si ciertos síntomas como debilidad muscular grave (el niño ya no puede levantarse de la cama, disfagia grave), deterioro de la función miocárdica o pulmonar o úlceras cutáneas indican un curso grave, debe añadirse ciclofosfamida. Si no hay mejoría, deben añadirse inmunoglobulinas intravenosas o ciclosporina A, o cambiar a biológicos (rituximab, infliximab, adalimumab). Desde un punto de vista pediátrico, el aumento de la calcinosis del tejido subcutáneo o muscular (Figs. 3 y 4 ) es un signo de inflamación sistémica persistente, que justifica la intensificación de la terapia. También se han realizado con éxito trasplantes de células madre en pacientes con DMJ con un curso muy grave; algunos pacientes lograron una remisión completa sin terapia. Cuando se alcanza la remisión clínica, la calcinosis puede desaparecer por completo.
Con buenos resultados, esta terapia estándar sigue asociada al riesgo de efectos secundarios no deseados, como infecciones, debilidad muscular inducida por los esteroides o náuseas semanales con el metotrexato. En el futuro, los nuevos biomarcadores mencionados podrían permitir la identificación precoz de un nuevo brote de la enfermedad o una reducción más rápida de la terapia. Los biomarcadores podrían así apoyar el camino hacia una terapia personalizada para esta enfermedad rara.
| Herramienta de diagnóstico basada en la web Mientras tanto, existe una herramienta web que, además del diagnóstico, también ofrece una probabilidad del mismo con la clasificación del subgrupo. Estos criterios han sido validados tanto para niños como para adultos con sospecha de miopatía inflamatoria. http://www.imm.ki.se/biostatistics/calculators/iim/ |
Literatura:
- Piper CJM, et al: Las células B CD19(+)CD24(hi)CD38(hi) se expanden en la dermatomiositis juvenil y muestran un fenotipo proinflamatorio tras la activación mediante el receptor Toll-Like 7 y el interferón alfa. Front Immunol 2018; 9: 1372.
- Gowdie PJ, Allen RC, Kornberg AJ, Akikusa JD: Características clínicas y evolución de la enfermedad en pacientes con dermatomiositis juvenil. Int J Rheum Dis 2013; 16(5): 561-567.
- Rider LG, Nistala K: Las miopatías inflamatorias idiopáticas juveniles: patogenia, fenotipos clínicos y de autoanticuerpos, y resultados. J Intern Med 2016; 280(1): 24-38.
- Huber A, Feldman BM: Resultados a largo plazo en la dermatomiositis juvenil: ¿cómo hemos llegado hasta aquí y adónde vamos? Curr Rheumatol Rep 2005; 7(6): 441-446.
- Huber AM, et al: Validación preliminar y significado clínico de la Herramienta de Evaluación Cutánea en la dermatomiositis juvenil. Arthritis Rheum 2008; 59(2): 214-221.
- Enders FB, et al: Recomendaciones basadas en el consenso para el tratamiento de la dermatomiositis juvenil. Ann Rheum Dis 2017; 76(2): 329-340.
- Wienke J, et al: Galectina-9 y CXCL10 como biomarcadores de la actividad de la enfermedad en la dermatomiositis juvenil: un estudio longitudinal de cohortes y validación de múltiples cohortes. Arthritis Rheumatol 2019. doi.org/10.1002/art.40881
- Brown VE, et al: Una encuesta de consenso internacional sobre los criterios diagnósticos de la dermatomiositis juvenil (DMJ). Reumatología (Oxford) 2006; 45(8): 990-993.
- Lundberg IE, et al.: 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis 2017; 76(12): 1955-1964.
- Huber AM, et al: Validación e importancia clínica de la escala de evaluación de la miositis infantil para evaluar la función muscular en las miopatías inflamatorias idiopáticas juveniles. Arthritis Rheum 2004; 50(5): 1595-1603.
- Jain M, et al: Fiabilidad intrainstitucional e interinstitucional de la prueba muscular manual (MMT) de fuerza de 10 puntos en niños con miopatías inflamatorias idiopáticas juveniles (MIJI). Phys Occup Ther Pediatr 2006; 26(3): 5-17.
- Lazarevic D, et al: Los criterios PRINTO para la enfermedad clínicamente inactiva en la dermatomiositis juvenil. Ann Rheum Dis 2013; 72(5): 686-693.
- Mathiesen PR, et al: Aptitud aeróbica tras la DMJ: un estudio de seguimiento a largo plazo. Reumatología (Oxford) 2013; 52(2): 287-295.
PRÁCTICA DERMATOLÓGICA 2019; 29(4): 26-29