Un ama de casa de 54 años sufrió un episodio agudo de angioedema de la lengua y la pared faríngea posterior y requirió tratamiento de urgencia en la unidad de cuidados intensivos (Fig. 1). Como las hinchazones se produjeron 30 minutos después de comer espaguetis con salsa de tomate y almejas, se sospechó de un acontecimiento alérgico y se remitió a la paciente para una aclaración alergológica. El informe médico mencionaba una pancitopenia leve preexistente.
La historia alergológica fue improductiva en cuanto a enfermedades atópicas preexistentes; en particular, no había pruebas de reacciones alérgicas o de intolerancia previas en relación con los alimentos. En consecuencia, las pruebas de punción (cribado de atopia, resumen de alimentos, incluidos los mejillones) y las determinaciones de IgE fueron negativas. Antes del episodio, la paciente no tomaba preparados que contuvieran AAS ni antiinflamatorios no esteroideos, ni estaba en tratamiento con inhibidores de la ECA o antagonistas de la angiotensina (AT) II (sartanes) [1, 2].
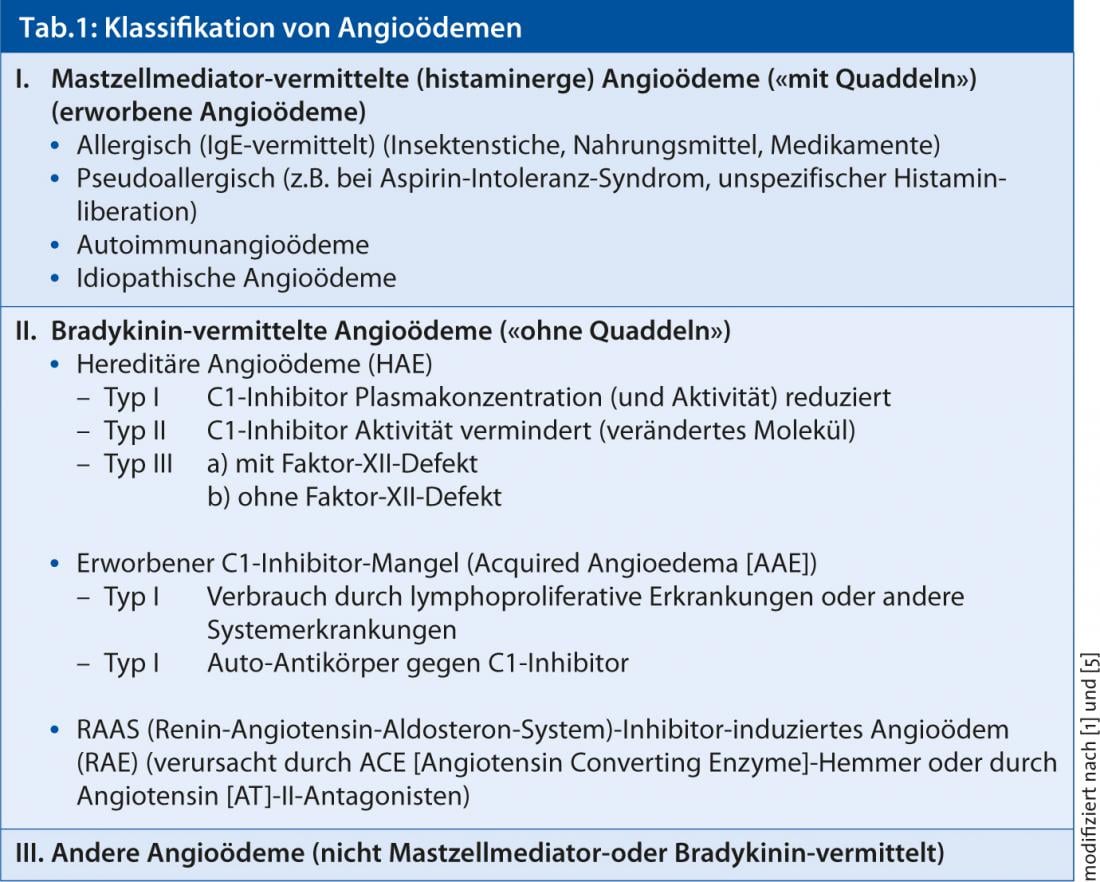
En la consideración diagnóstica diferencial del angioedema (tabla 1 ), en ausencia de pruebas anamnésicas de factores desencadenantes, el dolor abdominal que se queja ocasionalmente se interpretó en relación con la localización particular del episodio de angioedema en el sentido de un angioedema hereditario y se examinó el perfil del complemento.
Tanto la proteína inhibidora de C1 (<0,03 g/l; rango normal 0,2-0,36) como los niveles de C4 (<0,01 g/l; rango normal 0,1-0,4) no eran medibles. Del mismo modo, la función del inhibidor C1 era muy baja en <18% (norma 70-135%; Laboratorio Inmunológico, USZ).
Bajo el diagnóstico de trabajo de angioedema hereditario (tipo I), se inició una terapia con el andrógeno atenuado danazol (Danatrol), inicialmente 40 mg/día durante quince días, luego 200 mg/día durante otras dos semanas, para la profilaxis de las convulsiones. Un control de los parámetros de coagulación reveló la presencia de anticuerpos antifosfolípidos (anticardiolipina -IgM 520 UI/l; norma <15 UI/l); la serología para autoanticuerpos antinucleares y antiADN fue negativa, descartando el lupus eritematoso sistémico (LES). El diagnóstico original de angioedema hereditario se revisó a “angioedema adquirido [AAE] con deficiencia del inhibidor C1 en asociación con el síndrome antifosfolípido (APS)”.
El diagnóstico de AAE se confirmó posteriormente con la determinación de niveles de C1q y C1r disminuidos en dos ocasiones (C1q 0,014 g/l; norma: 0,0460-1116 g/l y C1r 37%; norma: 75-125%). Finalmente, una biopsia de médula ósea condujo al diagnóstico de linfoma no Hodgkin de células B (LNH, IgG κ).
Diagnóstico
Angioedema adquirido (AAE) en la deficiencia adquirida del inhibidor C1 (tipo I) debido al consumo/presencia de un inhibidor en un linfoma no Hodgkin.
Debate
En función de los mediadores implicados, el angioedema se divide ahora patogenéticamente en mediado por mastocitos o histaminérgico (a menudo acompañado de urticaria) y mediado por bradicinina (sin habones) (Tabla 1) [1, 5]. Entre estos últimos, además del angioedema hereditario (AEH) (tipos II y III) debido a la deficiencia del inhibidor C1 y el angioedema inducido por inhibidores del sistema renina-angiotensina-aldosterona (SRAA) causado por inhibidores de la ECA o antagonistas de la AT-II, existe otra forma extremadamente rara de angioedema que también se caracteriza por la deficiencia del inhibidor C1 de la esterasa (C1-INH) [4]. Sin embargo, la deficiencia en este caso no está causada por un cambio en los genes (factores hereditarios), sino por ciertas enfermedades o procesos “adquiridos” en el organismo que consumen o inactivan (o inhiben) cada vez más la C1-INH. La consecuencia: Hay muy poca C1-INH (funcional) disponible para frenar la formación excesiva de bradicinina. Los niveles de bradicinina aumentan y puede aparecer hinchazón en la piel y las mucosas. A diferencia del angioedema hereditario, esta rara forma de angioedema mediado por bradicinina suele aparecer después de los 30 años y no afecta a otros miembros de la familia. El angioedema adquirido puede ser el concomitante de otra enfermedad subyacente, que en cualquier caso debe ser aclarada por un médico.
En el caso que nos ocupa, inicialmente fue decisivo que se determinaran los factores del complemento C4 y el inhibidor C1 para excluir un angioedema histaminérgico, por ejemplo en caso de sospecha de alergia alimentaria. La determinación adicional de los primeros componentes del complemento C1q y C1r sirvió para diferenciar entre un AEH y un AAE. La enfermedad subyacente en la AAE tuvo que investigarse más a fondo serológicamente (exclusión de enfermedades autoinmunes, como el LES, etc.) y mediante biopsia de médula ósea [6]. La pancitopenia podría explicarse en el contexto de la afectación de la médula ósea del linfoma no Hodgkin.
Conclusión
En la mayoría de los casos, el angioedema se debe a causas alérgicas. Sin embargo, también existen angioedemas no causados por alergias, que se producen comparativamente en muy raras ocasiones y cuyo trasfondo médico a menudo pasa desapercibido durante un largo periodo de tiempo. Dado que los distintos tipos de angioedema requieren tratamientos diferentes, primero debe identificarse el motivo de la hinchazón.
Básicamente, se diferencian las siguientes formas de angioedema:
- Angioedema mediado por histamina (esto incluye tanto la alergia como el angioedema inducido por fármacos).
- Angioedema mediado por bradicinina o hereditario (AEH) causado por deficiencia o defecto del inhibidor C1.
Este caso clínico ha demostrado que la deficiencia del inhibidor C1 no sólo es hereditaria, sino que en raras ocasiones puede ser adquirida, en cuyo caso se denomina angioedema adquirido (AAE). El factor decisivo para el diagnóstico es el examen de laboratorio. La realización de pruebas de punción y la determinación de los inhibidores C1 y C4 permiten descartar el angioedema histaminérgico como primer paso. Midiendo los niveles de los valores sanguíneos individuales C1q y C1r, se puede distinguir entre AEH y AAE en un paso posterior. Mientras que la primera hinchazón
Mientras que los ataques de AEH siempre se producen antes de los 30 años, el raro AAE suele aparecer después de esta edad. Un AAE puede ser un indicador de una enfermedad subyacente y no debe ignorarse.
Literatura:
- Wüthrich B: Angioedema. Raramente alérgica. Primera parte: Clasificación, fisiopatología, diagnóstico. Foro Med Suiza 2012; 12 (7): 138-143.
- Wüthrich B: Angioedema. Raramente alérgica. Segunda parte: Terapia. Switzerland Med Forum 2012;12 (8): 175-178.).
- Wüthrich B: ¿Está presente el edema de Quincke? Diagnóstico diferencial con el angioedema. DERMATOLOGIE PRAXIS 2012; 1: 20.
- Wüthrich B: ¿Cuál es su diagnóstico? (Cuestionario). DERMATOLOGIE PRAXIS 2011;3: 38 y 42.
- Maurer M, Magerl M: JDDG 2010; 8/9: 663-672.
- Beretta KR, et al: Schweiz Med Wschr 1991; 121: 943-947.