El término linfomas indolentes engloba un número creciente de linfomas y leucemias no hodgkinianos poco malignos predominantemente de la serie de células B. Se diferencian y clasifican entre sí en función de su constelación de marcadores y su perfil genético. El tratamiento depende de la extensión o dinámica de la enfermedad y de los síntomas del paciente. Actualmente se está produciendo un cambio en la terapia, pasando de los quimioterapéuticos clásicos a los inhibidores de señales y los inmunoterapéuticos. El objetivo principal del tratamiento suele ser controlar la enfermedad durante el mayor tiempo posible con una tolerancia aceptable de la terapia.
El término linfoma no Hodgkin indolente (LNHi) engloba un grupo biológicamente heterogéneo de linfomas de células maduras y pequeñas de la serie de células B con tendencia a la manifestación leucémica. En el pasado, el término “indolente” describía un grupo de linfomas de crecimiento lento y “escasa malignidad” cuyos subtipos eran a menudo difíciles de separar entre sí y que no siempre necesitaban diferenciarse debido a la falta de opciones terapéuticas.

Entretanto, la ampliación de los datos inmunofenotípicos y moleculares permite una mejor subdivisión en entidades de linfoma, que debería realizarse de acuerdo con la actual clasificación de linfomas de la OMS de 2008 (Tab. 1) [1]. Hay iNHL que se transforman en linfomas blásticos o agresivos altamente malignos en el curso de la enfermedad, en primer lugar el linfoma folicular, seguido de la leucemia linfocítica crónica (LLC). Además, existen entidades como el linfoma de células del manto (LCM), la mayoría de las cuales (aprox. el 90% de todos los casos) muestran un comportamiento de crecimiento agresivo y se tratan mediante inmunoquimioterapia intensiva (si es necesario con terapia de altas dosis y sustitución autóloga de células madre). Pero también existe un subgrupo indolente que se manifiesta sobre todo en pacientes de edad avanzada con afectación de la médula ósea y el bazo. Sin olvidar los LNH típicos que pueden darse en un contexto clínico específico como el MALT asociado a Helicobacter pylori o el linfoma de la zona marginal asociado al VHC.
Diagnóstico y subtipos
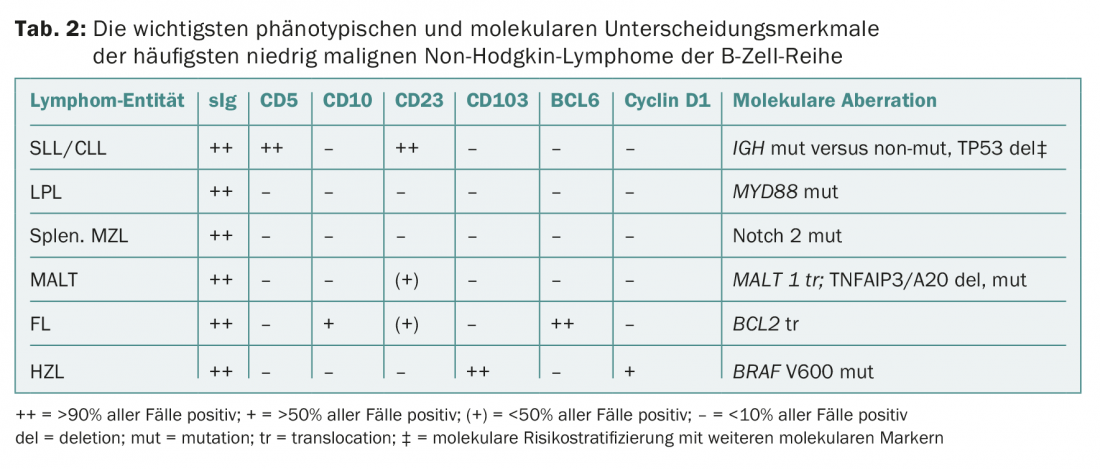
El diagnóstico se realiza mediante biopsia de los ganglios linfáticos y/o de la médula ósea, fenotipado (inmunológico) de la sangre y/o de la médula ósea y análisis genético-moleculares (Tab. 2) . Estas pruebas están estandarizadas y se aplican a todas las patologías de linfoma. Según la clasificación de la OMS, las siguientes entidades pertenecen al iNHL:
- Leucemia linfocítica crónica (LLC) resp. su forma ganglionar (linfoma linfocítico de células pequeñas, SLL)
- Linfoma linfoplasmocítico (LPL o enfermedad de Waldenström)
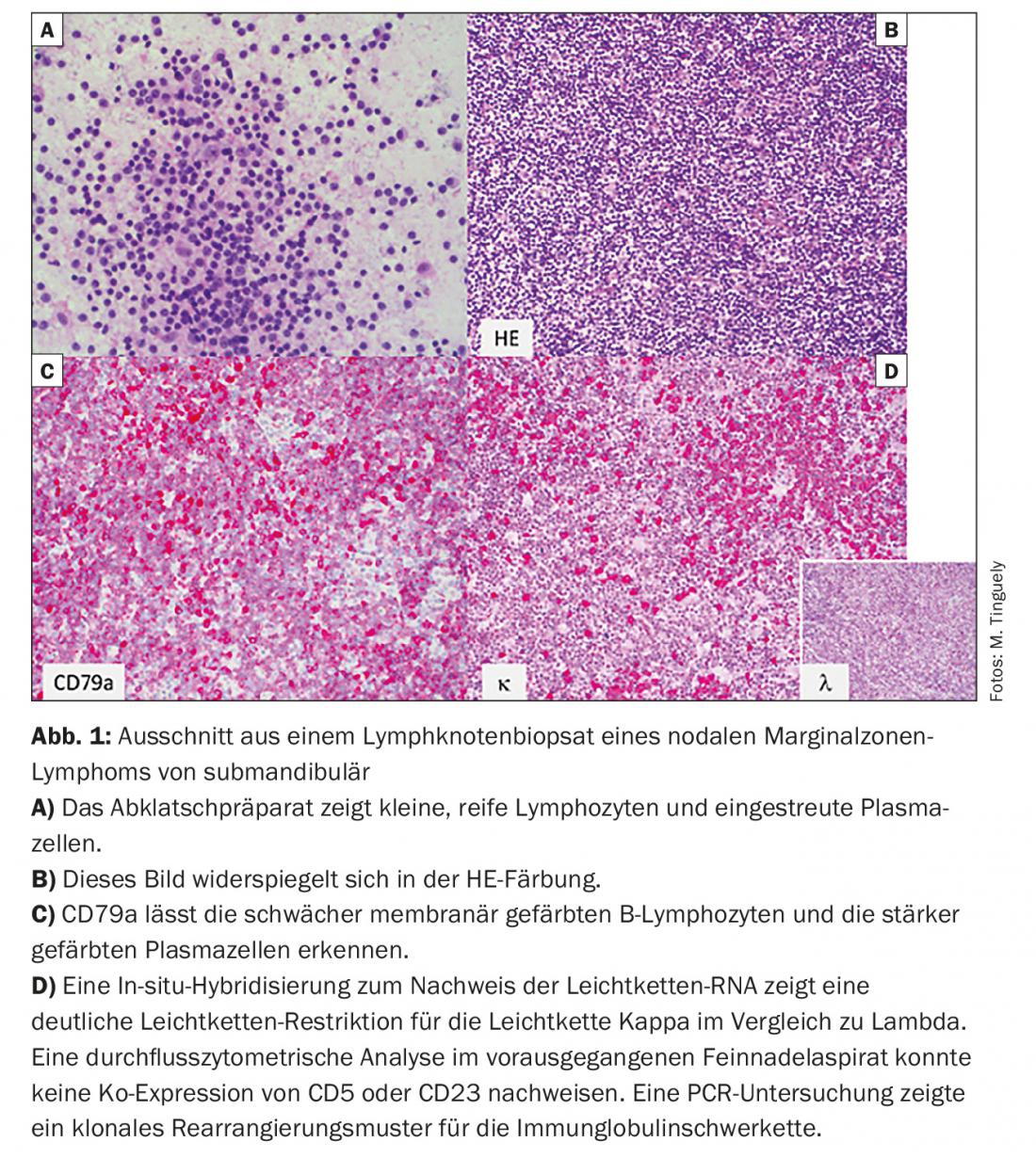
- Linfoma de la zona marginal (LZM) (Fig. 1) con sus tres subtipos: nodal, extranodal (por ejemplo, como MALT) y esplénico.
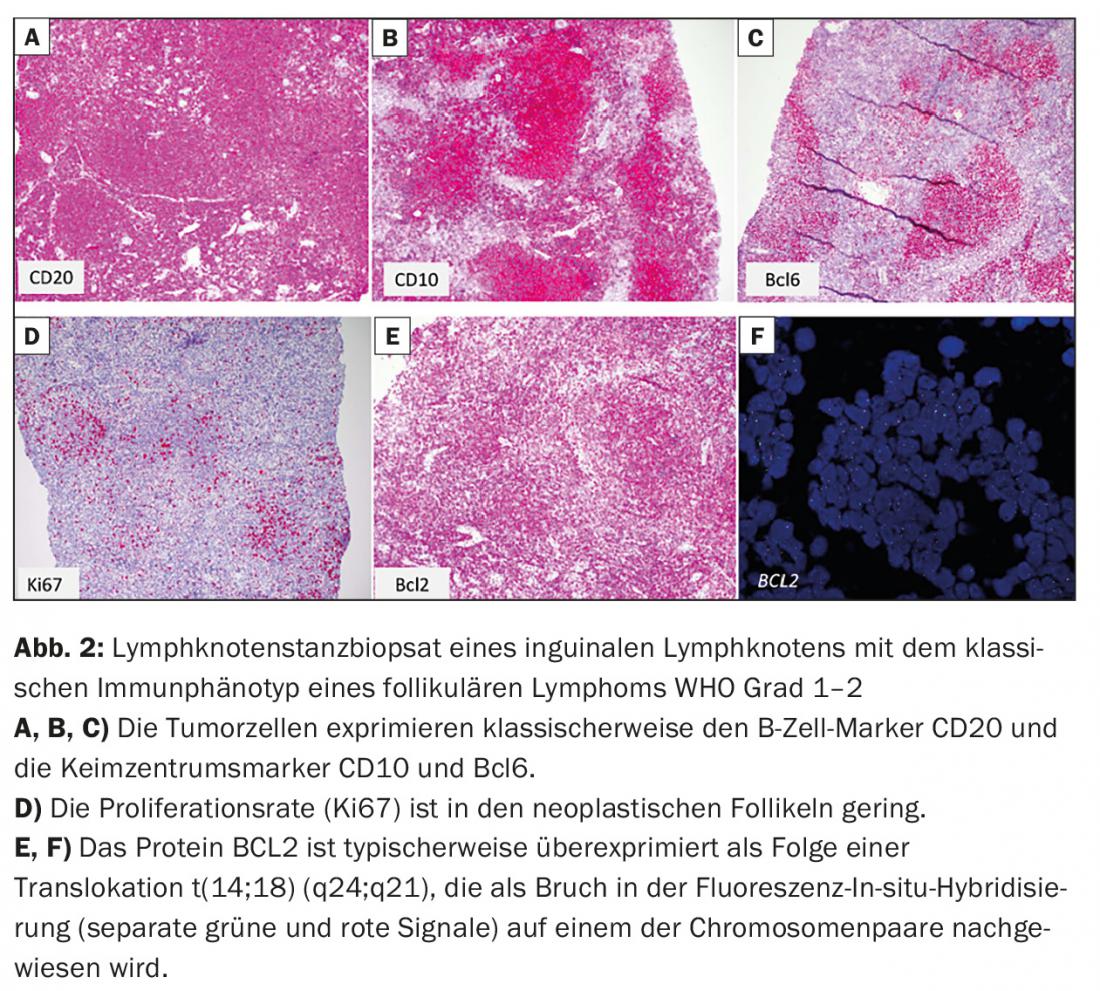
- Linfoma folicular (FL) (Fig. 2)
- Leucemia de células pilosas (HZL).
Todos los grupos de trabajo consideran que la LLC y la leucemia de células pilosas no forman parte del iNHL porque, como su nombre indica, suelen manifestarse principalmente con cambios en el recuento sanguíneo y en algunos casos (esto se aplica en particular a la leucemia de células pilosas) requieren otras medidas terapéuticas. El linfoma de células del manto (LCM) ya no debe contabilizarse como iNHL por las razones mencionadas, aunque existe un subgrupo indolente en pacientes de edad avanzada.
Presentación clínica y principios terapéuticos
Los linfomas indolentes suelen ser una enfermedad de edad avanzada. La gran mayoría de los pacientes (66%) ya se encuentran en estadios avanzados III y IV (estadificación según la clasificación de Ann Arbor o, más recientemente, la de Lugano) en el momento del diagnóstico y, por tanto, ya no son susceptibles de terapia curativa [2]. El diagnóstico no siempre va asociado a la necesidad de iniciar una terapia de inmediato. Aunque las recomendaciones varían un poco en función de cada subtipo, sigue siendo cierto que sólo debe tratarse la enfermedad sintomática. La definición de enfermedad sintomática ha permanecido prácticamente inalterada durante los últimos 30 años e incluye los síntomas locales debidos al crecimiento del linfoma, el deterioro de la función normal de los órganos (por ejemplo, anemia sintomática), los síntomas B, la enfermedad extraganglionar sintomática, otras citopenias o una rápida tasa de crecimiento de una manifestación del linfoma. Estos criterios son muy elásticos y ofrecen al paciente y al médico un amplio margen de decisión. Por lo tanto, actualmente se están realizando esfuerzos para establecer una mejor clasificación pronóstica y, por lo tanto, una ayuda a la toma de decisiones mediante nuevos marcadores (moleculares). Un ejemplo es el índice pronóstico de la LLC presentado recientemente (CLL-IPI), que proporciona una recomendación sobre el momento y el tipo de terapia [3].
Radioterapia en fases tempranas: ¿una opción curativa?
La radioterapia se ha definido en las últimas décadas como una opción de tratamiento curativo para los estadios iniciales del linfoma folicular. El 15-25% de todos los pacientes diagnosticados de linfoma folicular presentan un estadio I o II y las directrices nacionales e internacionales actuales siguen recomendando la radioterapia para estos pacientes [4].
La dificultad de una recomendación positiva para la aplicación de la radioterapia en la rutina clínica actual se basa en la incertidumbre de si los datos de los estudios antiguos (superficies de irradiación de gran tamaño, dosis totales elevadas, aplicaciones no específicas para los ganglios linfáticos o los campos involucionados) pueden transferirse a las técnicas de irradiación actuales. Esto plantea el gran dilema de que actualmente somos incapaces de definir el supuesto valor curativo de la radioterapia para los linfomas indolentes (principalmente el linfoma folicular) en las fases iniciales.
La espera tras el diagnóstico: ¿sigue siendo válida?
Algunos autores suponen que actualmente alrededor del 50% de los pacientes aún no necesitan tratamiento inmediato en el momento del diagnóstico. El término “vigilar y esperar” (watch & wait, w&w) se acuñó hace más de 35 años [5]. La espera parecía justificable cuando la enfermedad progresaba lentamente y no había síntomas o éstos eran leves, y también porque no existían terapias eficaces. En ese momento, la mediana de tiempo hasta la primera terapia para los pacientes con FL era de 31-36 meses. Los análisis observacionales longitudinales mostraron que alrededor del 20% de los pacientes con FL no requirieron terapia en una mediana de seguimiento de 17 años. En comparación con una cohorte de pacientes tratados con quimioterapia en el momento del diagnóstico, no hubo diferencias en la supervivencia global (SG) a 5 años. La mediana de la SG fue de once años y varió mucho entre las distintas histologías.
La idoneidad del w&w afecta a todos los iNHL, pero se analiza principalmente en el FL. Así, en un estudio multicéntrico, 379 pacientes fueron distribuidos aleatoriamente en tres brazos de tratamiento: sólo observación (w&w), sólo inducción con rituximab (aplicaciones cada cuatro semanas) o inducción con rituximab seguida de dos años de terapia de mantenimiento (rituximab cada dos meses) [6]. Esto mostró una ventaja significativa en la supervivencia libre de progresión (SLP) para ambos brazos de rituximab en comparación con w&w. Sin embargo, la supervivencia global no difirió, por lo que aún hoy no existe una razón de peso para el uso precoz de la inmunoterapia. Esto puede cambiar con las nuevas terapias (inmunológicas) y, por lo tanto, debe cuestionarse siempre.
Terapias actuales
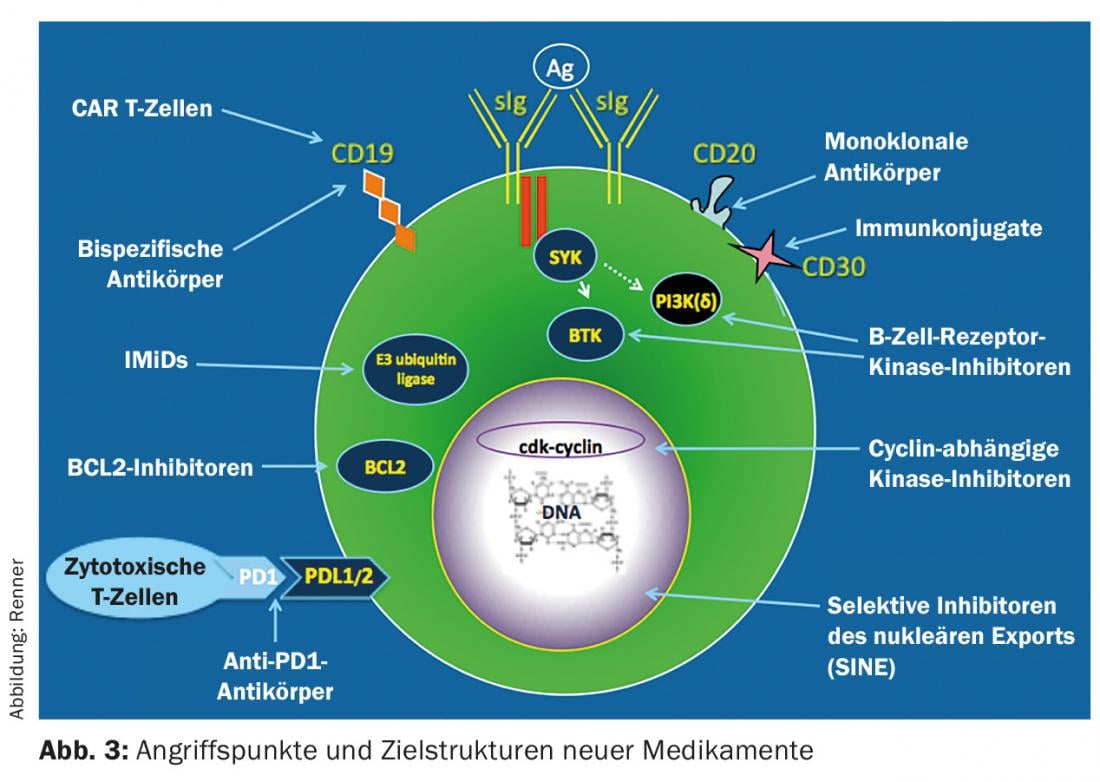
Las opciones de tratamiento para el iNHL han cambiado fundamentalmente en los últimos años con el establecimiento de nuevas terapias. Éstas se basan principalmente en anticuerpos y en la inhibición de la tirosina quinasa (Fig. 3 ). Hasta ahora, la inmunoterapia con anticuerpos CD20-específicos como monoterapia o en combinación con agentes quimioterapéuticos clásicos ha dominado la terapia de primera línea. Hay que seguir asumiendo que la curación no es posible a pesar de los métodos terapéuticos modernos; la única excepción es el trasplante alogénico de células madre.
Anticuerpos monoclonales específicos de CD20
Un ejemplo clásico de anticuerpo CD20-específico es el rituximab, que en la actualidad forma parte integral del tratamiento del linfoma de células B y puede utilizarse para diversas entidades como monoterapia (por ejemplo, FL) o en combinación con quimioterapia (por ejemplo, CLL). Los anticuerpos anti-CD20 más recientes (por ejemplo, el obinutuzumab) tienen una toxicidad celular directa aún mayor (actividad ADCC) y, por tanto, pueden eliminar las células del linfoma de forma más eficaz [7]. En el tratamiento de la LLC, por ejemplo, esto conlleva un aumento de los pacientes con enfermedad mínima residual (EMR) negativa. Esto significa que en los pacientes con obinutuzumab + quimioterapia, la enfermedad – en comparación con el tratamiento convencional con rituximab + quimioterapia – es más a menudo indetectable (del 38 al 3% en sangre periférica), a pesar de los métodos de detección sensibles. Queda por ver si el aumento de la tasa de negatividad de la ERM también conducirá a una supervivencia prolongada.
Combinación de anticuerpos y quimioterapia
Normalmente, los anticuerpos CD20-específicos se utilizan en combinación con clorambucilo, bendamustina o CHOP. El clorambucil se utiliza a menudo en combinación con obinutuzumab en el tratamiento de pacientes con LLC de edad avanzada o rituximab en el linfoma MALT [8]. Por otro lado, la bendamustina, en combinación con rituximab, se considera el estándar de primera línea en el linfoma folicular (grados 1 y 2) [9] y la LLC (con reservas sobre la fludarabina-endoxan). El rituximab con CHOP se utiliza con algo menos de frecuencia que en el pasado y se emplea principalmente para las variantes de linfoma blastoide o FL de grado 3 . En general, el número de ciclos de quimioterapia suele limitarse a seis (cada tres o cuatro semanas) y la inmunoterapia se administra durante el mismo tiempo o en monoterapia durante dos años como terapia de mantenimiento.
Inmunomoduladores
Los inmunomoduladores (IMID) son pequeñas moléculas que suelen tomarse por vía oral. Los IMID como la lenalidomida se utilizaron por primera vez con éxito en el mieloma múltiple, y ahora también se están probando en el linfoma con tasas de respuesta alentadoras. La combinación de lenalidomida y rituximab en pacientes con FL de grado 3 y enfermedad recidivante o refractaria logró una tasa de respuesta global (ORR) de hasta el 86%. Cuando la combinación se utiliza directamente en la terapia de primera línea, pueden alcanzarse ORR de hasta el 98% con altas tasas de remisión completa (RC) (87% de RC y RC no confirmada) y negatividad de MRD. Recientemente, también se concedió la aprobación para el tratamiento del linfoma de células del manto (LCM). La aprobación se basa en estudios con enfermedad MCL en recaída o refractaria debido a una ORR del 42-53% [10,11].
Inhibición de la vía de señalización del receptor de células B (BCR)
Pueden conseguirse tasas de respuesta aún mayores en el tratamiento de la MCL con compuestos que inhiban la vía de señalización descendente del BCR, por ejemplo, los inhibidores de la tirosina quinasa de Bruton (BTK) o de la PI3 quinasa (PI3K) [12]. Ambas quinasas son a menudo constitutivamente activas en las células de linfoma y promueven la proliferación o la supervivencia celular.
Inhibidores de la BTK
El ibrutinib es el primer inhibidor de la BTK aprobado que se une de forma irreversible y covalente a un residuo de cisteína (Cys-481) de la tirosina cinasa, provocando una inhibición fuerte y sostenida de la actividad enzimática. En la LLC, el ibrutinib mostró una elevada actividad. La aprobación se basa en un estudio comparativo con el anticuerpo monoclonal CD20-específico ofatumumab (estudio RESONATE) en 391 pacientes con LLC/SLL pretratados [13]. En una mediana de seguimiento de 9,4 meses, el ibrutinib (420 mg/d/po) mejoró significativamente la SLP y la SG. Después de doce meses, la SG fue del 90% en el grupo de ibrutinib y del 81% en el grupo de ofatumumab. La tasa de respuesta global fue significativamente mayor para el ibrutinib (42,6 frente a 4,1%, p <0,001). La tasa de respuesta y la duración fueron independientes de la presencia de del17p o de la resistencia a los análogos de las purinas. Esto demuestra el gran valor de los inhibidores de la BTK en este subgrupo de pacientes con LLC difíciles de tratar. Los efectos adversos más frecuentes fueron diarrea, fatiga, fiebre y náuseas.
La segunda entidad aprobada en Suiza se refiere al tratamiento de la LCM recidivante. La base para la aprobación fue un ensayo de fase II multicéntrico de un solo brazo con 111 pacientes con LCM pretratados con una dosis de 560 mg de ibrutinib una vez al día. La publicación completa informa de una ORR del 66% con una tasa de RC del 17% y una mediana de duración de la respuesta (DOR) de 17,5 meses [4]. Curiosamente, la tasa de respuesta aumentó de forma continua en el curso del tratamiento (la denominada “respuesta incremental bajo tratamiento”), de modo que -a diferencia de las quimioterapias clásicas- también pueden producirse remisiones tardías con una terapia continuada.
Inhibidores de PI3K
La familia PI3K está formada por una serie de serina/treonina quinasas que regulan el crecimiento, la diferenciación, el metabolismo, la supervivencia y la proliferación en diversas células. La inhibición de la unidad p110δ, por ejemplo, conduce a una depleción significativa de las células B y al bloqueo de la vía de señalización descendente del BCR. Por ello, la mayoría de los enfoques terapéuticos en el tratamiento del linfoma se centran en el bloqueo directo de la unidad p110δ. El prototipo de esta clase de sustancias es el idelalisib como inhibidor selectivo de p110δ.
El idelalisib se probó inicialmente en un ensayo aleatorizado de fase III en 220 pacientes con LLC recidivante en combinación con rituximab [15]. En el brazo de control, los pacientes recibieron rituximab más placebo. Con una mediana de pretratamiento con tres sustancias, en casi todos los casos se había utilizado previamente rituximab y un alquilano o un análogo de nucleótidos de purina. Casi el 40% de los pacientes con LLC también presentaban la alteración genética desfavorable del gen p53.
En cuanto a la eficacia, hubo una tasa de respuesta significativamente mayor en el brazo de rituximab + idelalisib (81 frente a 13%), una prolongación significativa de la SLP a 1 año (93 frente a 46%, p<0,001) y una prolongación significativa de la SG a 1 año (92 frente a 80%, p=0,02). Se demostró la superioridad de la combinación de rituximab + idelalisib en todos los subgrupos.
Los efectos secundarios importantes para la práctica clínica diaria y que van más allá de la aplicación del rituximab son la diarrea precoz o, a veces, tardía. Sin embargo, si se compara el perfil de efectos secundarios con otras sustancias que podrían utilizarse en esta situación, como el ofatumumab, el alemtuzumab o los fármacos citostáticos convencionales, el idelalisib obtiene sin duda una puntuación positiva.
Como agente en monoterapia, el idelalisib muestra una gran eficacia en el iNHL en el contexto de la terapia de recaída. La sustancia está aprobada para el tratamiento de pacientes con FL con enfermedad recidivante que hayan recibido dos líneas previas de terapia [16]. En el ensayo subyacente de fase II de un solo brazo en 125 pacientes con iNHL resistentes a rituximab y alquilanos, se administró idelalisib a 150 mg dos veces al día hasta la progresión de la enfermedad. La mediana del tiempo hasta la respuesta fue de 1,9 meses, la mediana de la duración de la respuesta fue de 12,5 meses y la mediana de la SLP fue de 11 meses. Los acontecimientos adversos de grado 3 o superior más frecuentes fueron neutropenia (27% de los pacientes), elevación de las aminotransferasas (13%), diarrea (13%) y neumonía (7%).
Inhibidores de BCL-2
La proteína antiapoptótica BCL-2 se sobreexpresa en las células de linfoma y contribuye a la resistencia a la quimioterapia. Los inhibidores selectivos de BCL-2 administrados por vía oral, como el venetoclax, detienen la proliferación de las células de linfoma y provocan así remisiones tumorales en modelos preclínicos. En un ensayo clínico inicial de 106 pacientes con LCM (n=28), linfoma folicular (n=29), linfoma difuso de células B grandes (n=41) y otros subtipos de LNH (n=8), la monoterapia con venetoclax mostró un perfil de seguridad aceptable con la dosis máxima tolerada de 1200 mg/d [17]. La ORR fue del 44% para todos los subtipos, del 78% para el MCL y del 38% para el FL. La mediana de la SLP fue de 10-14 meses. Los acontecimientos adversos relacionados con el tratamiento (EA ≥20%) más frecuentes fueron náuseas (48%), diarrea (44%), fatiga (41%), disminución del apetito (21%) y vómitos (21%). De importancia es la aparición del síndrome de lisis tumoral, que, sin embargo, se produjo en dos pacientes sin consecuencias clínicas.
Terapias futuras
Las futuras terapias procederán principalmente de dos áreas (Fig. 3): Inhibidores de las vías de señalización que se dirigen a moléculas de conmutación importantes de la célula del linfoma y bloquean su función (por ejemplo, inhibidores CDK 4/6), y nuevos inmunoterapéuticos. La constatación de que el sistema inmunitario contribuye al control de los tumores está revolucionando actualmente las opciones de tratamiento hemato-oncológico. Los inhibidores del bloqueo de los puntos de control, los anticuerpos biespecíficos y las células T reprogramadas (células T CAR) se encuentran en fase de desarrollo clínico con algunos resultados impresionantes. Quizá algún día consigamos estimular el sistema inmunitario de tal forma que sea posible el control de los tumores a largo plazo e incluso su curación.
Literatura:
- Swerdlow SH, et al.: IARC Press, Lyon, 2008.
- Brice P, et al: J Clin Oncol 1997; 15(3): 1110-1117.
- Molica S, et al: Resumen 498, presentado en la 57ª Reunión Anual de la Sociedad Americana de Hematología (ASH), 2015.
- Hiddemann W, et al: Internist (Berl) 2016; 57(3): 222-229.
- Morrison VA, Peterson BA: Leuk Lymphoma 1993; 10 Sup: 29-33.
- Adreshna KM, et al: Lancet Oncol 2014; 15(4): 424-435.
- Goede V, et al: NEJM 2014; 370(12): 1101-1110.
- Zucca E, et al: J Clin Oncol 2013; 31(5): 565-572.
- Rummel MJ, et al: Lancet 2013; 381(9873): 1203-1210.
- Habermann TM, et al: Br J Haematol 2009; 145(3): 344-349.
- Witzig TE, et al: Ann Oncol 2011; 22(7): 1622-1627.
- Mato A, et al: Am J Hematol 2015; 90(7): 657-664.
- Byrd JC, et al: NEJM 2014; 371(3): 213-223.
- Wang ML, et al: NEJM 2013; 369(6): 507-516.
- Furman RR, et al: NEJM 2014; 370(11): 997-1007.
- Gopal AK, et al: NEJM 2014; 370(11): 1008-1018.
- Roberts AW, et al: NEJM 2016; 374(4): 311-322.
InFo ONCOLOGÍA Y HEMATOLOGÍA 2016; 4(2): 11-15