En hematología, la reunión anual de la Asociación Europea de Hematología es una de las citas del calendario anual. La asociación promueve la excelencia en la atención al paciente, la investigación y la educación en hematología. En consecuencia, el congreso transmite los últimos y más innovadores hallazgos y resultados de la investigación sobre las enfermedades hematológicas. Abarcará la investigación y la práctica clínicas, la investigación básica y traslacional, y los últimos enfoques para el diagnóstico y el tratamiento.
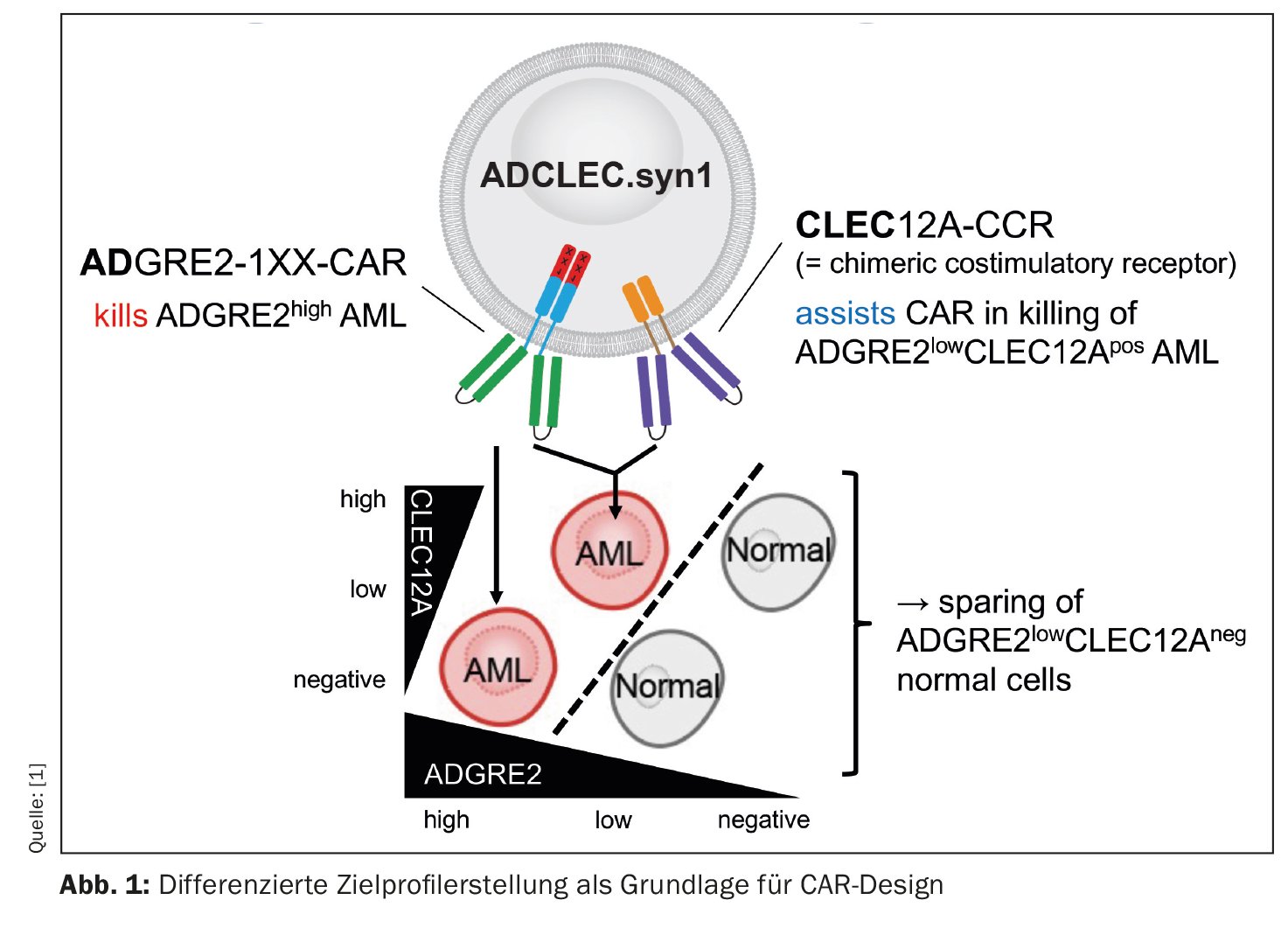
Un grupo de investigadores está investigando una nueva terapia CAR para la leucemia mieloide aguda (LMA) cuyos resultados preclínicos son alentadores [1]. El concepto denominado ADCLEC.syn1 utiliza receptores cooperativos dirigidos a ADGRE2 y CLEC12A. De este modo, se eliminará la LMA y se minimizarán al mismo tiempo las toxicidades hematológicas. Así lo ha demostrado una amplia serie de modelos de eficacia y toxicidad in vivo. Las terapias CAR para la LMA se enfrentan a obstáculos debidos a la heterogeneidad clonal y a la similitud con la hematopoyesis temprana normal, que pueden dar lugar a escapes de antígenos y toxicidades hematológicas. El presente estudio investigó la expresión cuantitativa de las dianas de superficie en la LMA y los tejidos normales para determinar las ventanas terapéuticas que pueden ser explotadas por novedosos diseños combinatorios de CAR. Así, se desarrolló ADCLEC.syn1, una novedosa terapia combinatoria CAR que se dirige conjuntamente a ADGRE2 y CLEC12A para la eliminación selectiva de las células de la LMA con bajos niveles de ADGRE2 y la preservación de las células madre y progenitoras hematopoyéticas normales. (Fig. 1). Los investigadores correlacionaron la expresión del antígeno diana con la eficacia de las células T ADCLEC.syn1 y CD33-CAR utilizando xenoinjertos de LMA. Los resultados mostraron que el ADCLEC.syn1 inducía una remisión duradera en varias líneas celulares de LMA humana representativas de los fenotipos de los pacientes con LMA en recaída/refractaria. Sin embargo, los ratones que habían recibido injertos de LMA y habían sido reconstituidos con células hematopoyéticas humanas normales sólo respondieron al ADCLEC.syn1 pero no al CD33-CAR. Estos resultados ponen de relieve la importancia del perfil cuantitativo de dianas de CAR en la LMA. ADCLEC.syn1 se está investigando ahora en un primer ensayo clínico de fase I en humanos para la LMA en recaída/refractaria.
Progresos en APL
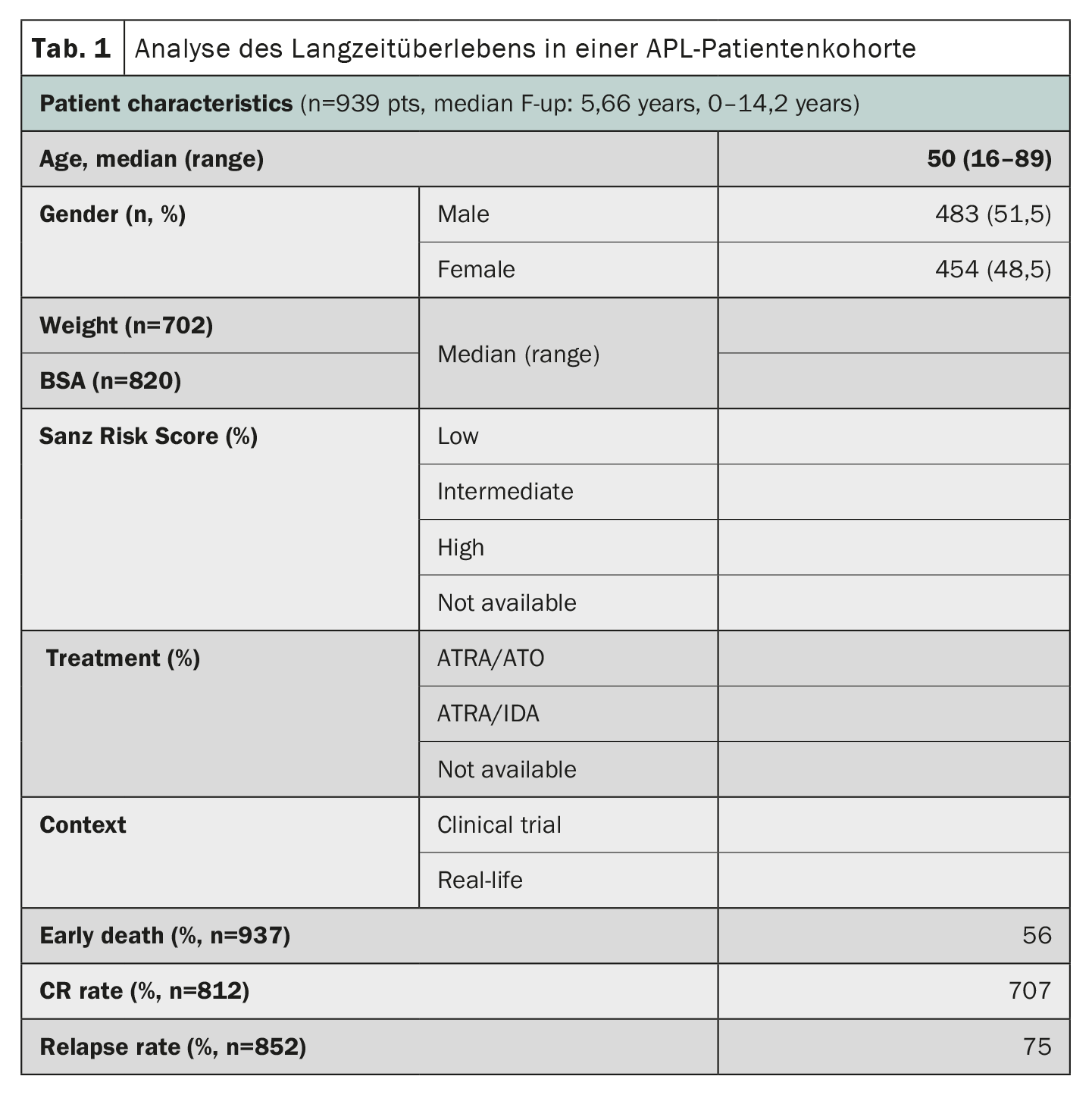
La leucemia promielocítica aguda (LPA), considerada en su día una de las formas de leucemia mieloide aguda más rápidamente mortales, ha mostrado notables progresos en los resultados de su tratamiento. Un estudio que utilizó datos del registro HARMONY con una gran cohorte de pacientes confirmó que la terapia combinada de ácido transretinoico total (ATRA) y trióxido de arsénico (ATO) dio lugar a tasas de supervivencia global a 10 años del 85-92% en pacientes con LPA [2]. El registro HARMONY incluye a 1868 pacientes con LPA, procedentes de dos ensayos clínicos y registros nacionales de seis países, diagnosticados entre 2007 y 2020. De ellos, 937 pacientes cumplieron los requisitos de calidad de los datos y se incluyeron en el presente análisis. Los datos se armonizaron utilizando un modelo de datos comunes de la Observational Medical Outcomes Partnership y se registraron en la plataforma de macrodatos HARMONY. Los resultados del análisis mostraron que los pacientes tratados con el régimen ATRA-ATO tenían una tasa de supervivencia global (SG) a 10 años del 92% en comparación con el 75% de los pacientes tratados con el régimen ATRA-idarubicina (AIDA) (Tab. 1). El beneficio para la supervivencia fue el mismo en los distintos grupos de riesgo definidos por la puntuación de riesgo de Sanz. La edad también desempeñó un papel importante en la supervivencia, y los pacientes más jóvenes (<50 años) obtuvieron mejores resultados. Sin embargo, la tasa de muertes tempranas (<30 días después del diagnóstico) fue similar en ambos grupos (3,4%-5,7%). En conjunto, estos resultados en una gran cohorte internacional de pacientes confirman el importante beneficio para la supervivencia de la terapia ATRA-ATO sin quimioterapia para los pacientes con LPA, independientemente de su perfil de riesgo, y aportan valiosas ideas para el tratamiento de la LPA.
¿Curación funcional en la TDT?
Los resultados provisionales iniciales son alentadores [3]: Un ensayo de fase III de autotemcel exagamglogénico (exa-cel), una terapia celular no vírica, está alimentando la esperanza de una cura funcional única para los pacientes con β-talasemia dependiente de transfusión (TDT). Se demostraron resultados significativos en la independencia de las transfusiones, la mejora de los niveles de hemoglobina y la calidad de vida. Exa-cel reactiva la síntesis de hemoglobina fetal (HbF) mediante la edición genética ex vivo CRISPR/Cas9, dirigida al gen BCL11A en células madre y progenitoras hematopoyéticas CD34+ autólogas. De los 48 pacientes con TDT que recibieron exa-cel, 27 fueron evaluables para los criterios de valoración del estudio en el análisis intermedio preespecificado. Los que fueron independientes de las transfusiones durante ≥12 meses tuvieron un tiempo medio hasta la última transfusión de 37 días tras la infusión de exa-cel y permanecieron libres de transfusiones durante 12,1-40,7 meses. Los niveles de hemoglobina, así como los alelos BCL11A editados en las células CD34+ de la médula ósea y en las células sanguíneas nucleadas periféricas, permanecieron estables a lo largo del tiempo.
El estudio también observó mejoras significativas en la calidad de vida y un injerto satisfactorio de neutrófilos y plaquetas en todos los pacientes, lo que pone de relieve la eficacia de la terapia. El perfil de seguridad de exa-cel fue coherente con el régimen de acondicionamiento mieloablativo basado en busulfán y los procedimientos de trasplante autólogo, con acontecimientos adversos manejables. Todos los acontecimientos adversos graves se resolvieron y no hubo muertes, retiradas del estudio ni neoplasias.
Eritrocitosis incontrolada en la policitemia vera
El estudio REVIVE investigó la eficacia de la rusfertida, un nuevo mimético de la hepcidina, en pacientes con policitemia vera (PV) [4]. La rusfertida inhibe la producción de glóbulos rojos en los pacientes con PV al limitar la disponibilidad de hierro. El estudio utilizó una proteína sintética similar a la hepcidina, que normalmente produce el hígado y regula el transporte de hierro, para tratar la eritrocitosis asociada a la PV (producción excesiva de glóbulos rojos). La fase de retirada aleatoria de 12 semanas del estudio cumplió el criterio de valoración principal y demostró la gran eficacia de la rusfertida para controlar la eritrocitosis, una característica clave de la PV que aumenta el riesgo de complicaciones tromboembólicas y cardiovasculares.
El estudio de fase II reclutó a pacientes diagnosticados de PV según los criterios de la OMS de 2016 y que requirieron un número excesivo de flebotomías terapéuticas (PT) en pacientes tratados con PT sola o con agentes de citorreducción (CYTO). La rusfertida, administrada por vía subcutánea una vez a la semana, se añadió a la terapia PV anterior. Durante la fase de retirada, se asignó aleatoriamente a los pacientes a continuar el tratamiento con rusfertida durante 12 semanas o a recibir placebo. Los datos de la fase de retirada aleatoria mostraron la eficacia superior de la rusfertida en comparación con el placebo. Los pacientes mostraron una tasa de respuesta estadísticamente significativa (sin TP) del 69,2% frente al 18,5% del grupo placebo. Además, el tratamiento con rusfertida se asoció a un control sostenido del hematocrito (HCT) en comparación con el placebo. La tasa de pacientes libres de TP alcanzó el 92,3%. En general, el tratamiento fue bien tolerado, y la mayoría de los acontecimientos adversos fueron reacciones en el lugar de la inyección de gravedad leve a moderada que disminuyeron con el tratamiento continuado.
Los resultados positivos demuestran la eficacia y tolerabilidad de la rusfertida como terapia altamente eficaz para la eritrocitosis incontrolada y los síntomas asociados en la PV y representan un avance significativo en el tratamiento de esta neoplasia mieloproliferativa maligna. El compuesto ofrece un enfoque novedoso basado en un mimético hormonal que actúa selectivamente sobre la eritrocitosis incontrolada, proporcionando un control sostenido y duradero de la HCT y una mejora de los síntomas relacionados con la PV.
Tratamiento de la anemia en LR-MDS
El ensayo de fase III COMMANDS mostró resultados prometedores en el tratamiento de la anemia asociada a los síndromes mielodisplásicos de bajo riesgo (LR-MDS) [5]. En un análisis provisional previamente planificado, el luspatercept mostró beneficios clínicos significativos en pacientes con SDM-LR sin AEE en comparación con el tratamiento estándar con epoetina alfa. Estos hallazgos tienen el potencial de cambiar el panorama del tratamiento de los pacientes con LR-MDS que dependen de las transfusiones. Los pacientes con LR-MDS que padecen anemia crónica sufren una mayor morbilidad, sobrecarga de hierro y una menor supervivencia. El tratamiento estándar actual, los agentes estimulantes de la eritropoyesis (AEE), sólo consiguen resultados subóptimos en estos pacientes. El análisis provisional evaluó la eficacia y seguridad de luspatercept y epoetina alfa en 356 pacientes con SDM-LR sin AEE y dependientes de transfusión. El criterio de valoración primario del estudio fue el logro de la independencia de la transfusión de glóbulos rojos (ITR) durante ≥12 semanas con un aumento simultáneo de la hemoglobina media de al menos 1,5 g/dL durante las primeras 24 semanas. El 59% de los pacientes tratados con luspatercept lograron un IT-RBC y un aumento concomitante de la hemoglobina en comparación con el 31% del grupo de epoetina alfa. Se observó un beneficio clínico en todos los subgrupos. La consecución de los criterios de valoración secundarios también fue favorable al luspatercept, incluida la mejora hematológica. Los pacientes con mutaciones genéticas específicas asociadas a los SMD, como SF3B1, SF3B1a, ASXL1 y TET2, respondieron mejor que la media al luspatercept, independientemente de su carga global de mutaciones. El luspatercept tuvo un perfil de seguridad favorable, con acontecimientos adversos relacionados con el tratamiento de leves a moderados. La tasa de mortalidad global fue similar en los grupos de luspatercept y epoetina alfa.
Tratar eficazmente la LLC
El estudio CLL14, una investigación sobre el tratamiento de la leucemia linfocítica crónica (LLC), ha proporcionado hallazgos recientes sobre los resultados a largo plazo de los pacientes tratados con venetoclax-obinutuzumab (Ven-Obi) [6]. Los resultados demuestran una eficacia y seguridad sostenidas y podrían establecer a Ven-Obi como la opción de tratamiento preferida para los pacientes con LLC, incluidos aquellos con enfermedad concomitante. En este estudio en curso, 432 pacientes con LLC no tratados previamente fueron asignados aleatoriamente al tratamiento con Ven-Obi o clorambucil-obinutuzumab (Clb-Obi). Tras una mediana de tiempo de seguimiento de 76,4 meses, Ven-Obi mostró una mejor supervivencia libre de progresión (SLP) que Clb-Obi (mediana de SLP 76,2 frente a 36,4 meses). Es importante destacar que, incluso después de seis años, la tasa de SLP estimada para Ven-Obi fue del 53,1% en comparación con el 21,7% para Clb-Obi. El estudio también demostró que Ven-Obi dio lugar a un tiempo hasta el siguiente tratamiento (TTNT) significativamente mayor en comparación con Clb-Obi (TTNT a 6 años del 65,2% frente al 37,1%) (Fig. 2). Estos resultados positivos se observaron en todos los grupos de riesgo, incluidos los pacientes con características de LLC de alto riesgo. Además, Ven-Obi mostró una excelente respuesta a la enfermedad mínima residual (EMR), con un 7,9% de pacientes sin niveles detectables de EMR cinco años después del tratamiento, en comparación con el 1,9% con Clb-Obi. No se detectaron nuevas señales de seguridad.
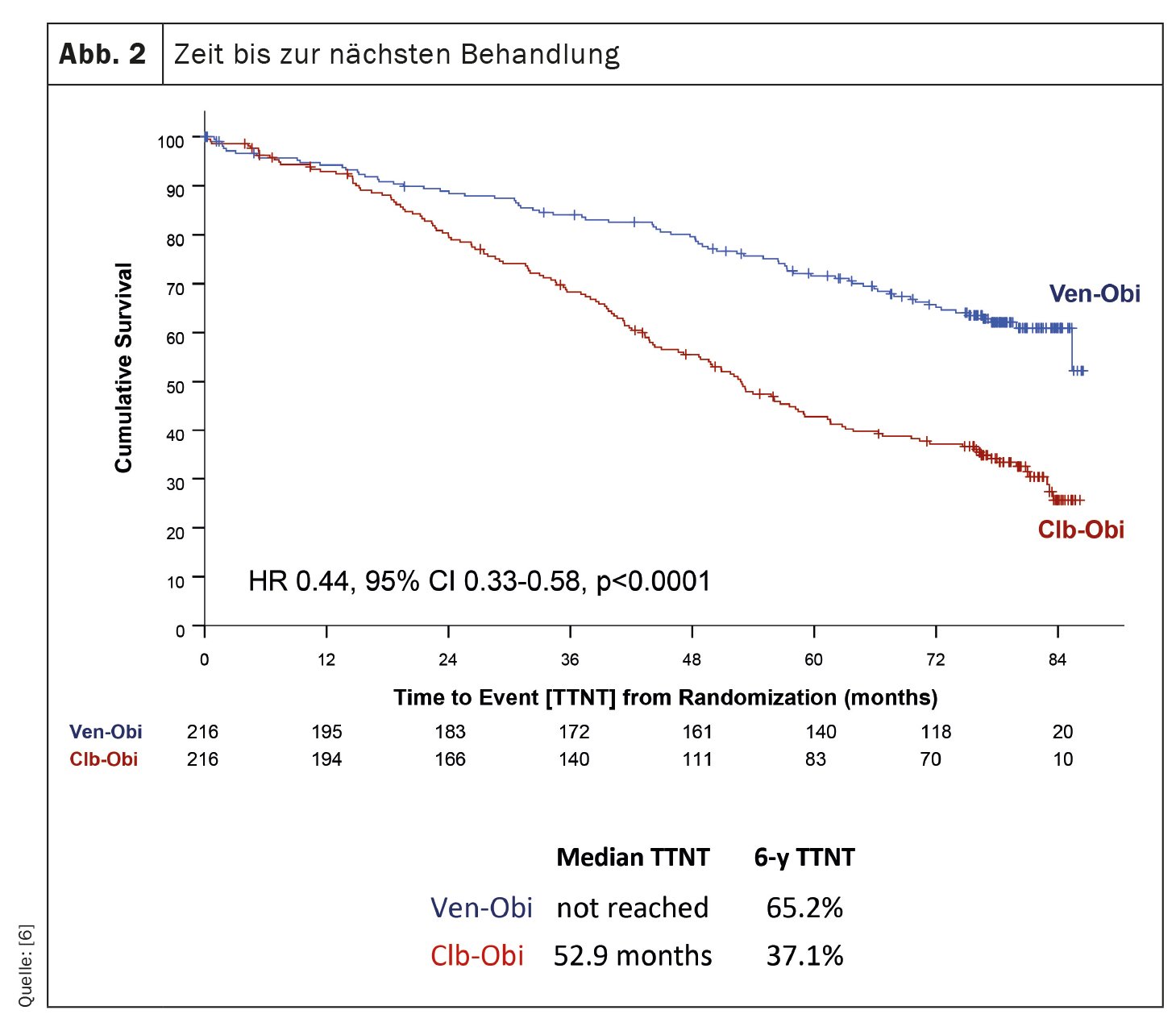
Ven-Obi ofrece una remisión sostenida, altas tasas de enfermedad mínima residual indetectable y un tiempo prolongado hasta el siguiente tratamiento. Más de la mitad de los pacientes siguen en remisión cinco años después de finalizar el tratamiento y la mayoría no necesita tratamiento de segunda línea.
Hemocromatosis hereditaria
La homocigosis de la variante HFE C282Y causa la hemocromatosis hereditaria, que puede provocar diabetes, enfermedades hepáticas y cardiacas. Un estudio ha comprobado ahora si la homocigosis C282Y aumenta el riesgo de diabetes, enfermedades hepáticas y cardiopatías, incluso en personas con un nivel normal de hierro plasmático, saturación de transferrina o ferritina [7]. También se investigó si los homocigotos C282Y con diabetes, enfermedades hepáticas o cardíacas tienen un mayor riesgo de muerte en comparación con los no portadores de estas enfermedades. Un total de 132 542 individuos consecutivos de una cohorte de población general fueron genotipados para la variante HFE C282Y y se encontraron 422 homocigotos. Se realizó un seguimiento prospectivo de los individuos durante un máximo de 27 años. La información sobre los contactos hospitalarios procedía del registro nacional de pacientes, en el que se inscriben todos los hospitales daneses.
Durante el periodo de seguimiento, 17.688 personas murieron por cualquier causa, mientras que 7.702, 2.804 y 21.769 personas fueron hospitalizadas por diabetes, enfermedades hepáticas y cardiopatías, respectivamente. Al comparar los homocigotos C282Y con los no portadores, los cocientes de riesgo fueron de 1,66 para la diabetes, 2,16 para la enfermedad hepática y 0,98 para la enfermedad cardiaca. El riesgo de diabetes aumentó incluso en los homocigotos C282Y con hierro, saturación de transferrina o ferritina normales (4,35; 1,81-10,48). Los homocigotos C282Y con diabetes tenían un cociente de riesgo de muerte por cualquier causa de 1,94 en comparación con los no portadores con diabetes. El porcentaje de todas las muertes en homocigotos C282Y que teóricamente podrían evitarse si se eliminara una sola enfermedad fue del 27,3% para la diabetes y del 14,4% para la enfermedad hepática. Los resultados sugieren que la estrategia actual de tratamiento de la hemocromatosis, centrada en reducir la ferritina, es insuficiente para reducir el riesgo de diabetes y el riesgo de morir por esta causa.
Congreso 2023 de la Asociación Europea de Hematología (EHA)
Literatura:
- Hauber S, et al.: Novel CAR Therapy for Acute Myeloid Leukemia Targeting ADGRE2 and CLEC12A Shows Favorable Pre-Clinical Outcomes. S104. 10.06.2023. EHA 2023.
- Voso MT, et al: El régimen ATRA-ATO proporciona una ventaja significativa en la supervivencia de los pacientes con leucemia promielocítica aguda. S136. 11.06.2023. EHA 2023.
- Locatelli F, et al.: Exagamglogene Autotemcel: A Potential One-Time Functional Cure for Patients with Transfusion-Dependent β-Thalassemia. S270. 11.06.2023. EHA 2023.
- Hoffman R, et al.: Rusfertide Therapy Serves as a Novel Effective Treatment for Uncontrolled Erythrocytosis in Polycythemia Vera. LB2710. 11.06.2023. EHA 2023
- Della Porta MG, et al.: Luspatercept Is Superior to Epoetin Alfa in Treating Anemia in Lower-Risk Myelodysplastic Syndromes. S102. 10.06.2023. EHA 2023.
- Al-Sawaf O, et al.: Long-Term CLL14 Study Confirms Venetoclax-Obinutuzumab as Effective Treatment for Chronic Lymphocytic Leukemia. S145. 09.06.2023. EHA 2023.
- Mottelson M, et al.: Individuals with HFE C282Y Homozygosity and Diabetes habe almost two-fold risk of death compared to non-carriers with diabetes: A prospective study of a 132,542-individual general population cohort. S280. EHA 2023.
InFo ONKOLOGIE & HÄMATOLOGIE 2023; 11(3): 26–28