El término colectivo “hipertensión pulmonar (HP)” engloba varios tipos de hipertensión pulmonar potencialmente mortales cuyos enfoques terapéuticos son muy diferentes y muy complejos. Dado que los síntomas son a menudo inespecíficos, incluso el diagnóstico es un gran desafío.
La clasificación clínica de la hipertensión pulmonar (HP) se divide actualmente en cinco grupos, como nos recordó al principio el Prof. Dr. Horst Olschewski, Jefe del Departamento Clínico de Neumología del Departamento Universitario de Medicina Interna del Hospital Universitario LKH de Graz:
- El grupo 1 incluye la hipertensión arterial pulmonar (HAP), la forma más rara de hipertensión pulmonar, que sólo se diagnostica tras descartar otras causas subyacentes.
- El grupo 2, hipertensión pulmonar en cardiopatía izquierda, es con diferencia la forma más común de hipertensión pulmonar.
- El grupo 3 incluye la hipertensión pulmonar debida a enfermedad pulmonar y/o deficiencia de oxígeno. Esta forma también se da con mucha más frecuencia que la PAH.
- El grupo 4 incluye la hipertensión pulmonar tras una embolia pulmonar, que afecta hasta al 4% de los pacientes que han sufrido una embolia pulmonar aguda.
- El grupo 5 incluye formas raras de hipertensión pulmonar que no pueden asignarse claramente a ninguno de los grupos mencionados, como la hipertensión pulmonar en la sarcoidosis o la enfermedad renal crónica.
Los grupos 2 y 3, según el Prof. Olschewski, representan juntos un total del 90% de toda la hipertensión pulmonar, lo que plantea peligros de diagnóstico diferencial.
Heterogeneidad del IPAH
Para el análisis de conglomerados, se identificó a los pacientes que cumplían los criterios de hipertensión arterial pulmonar idiopática (HAPI).
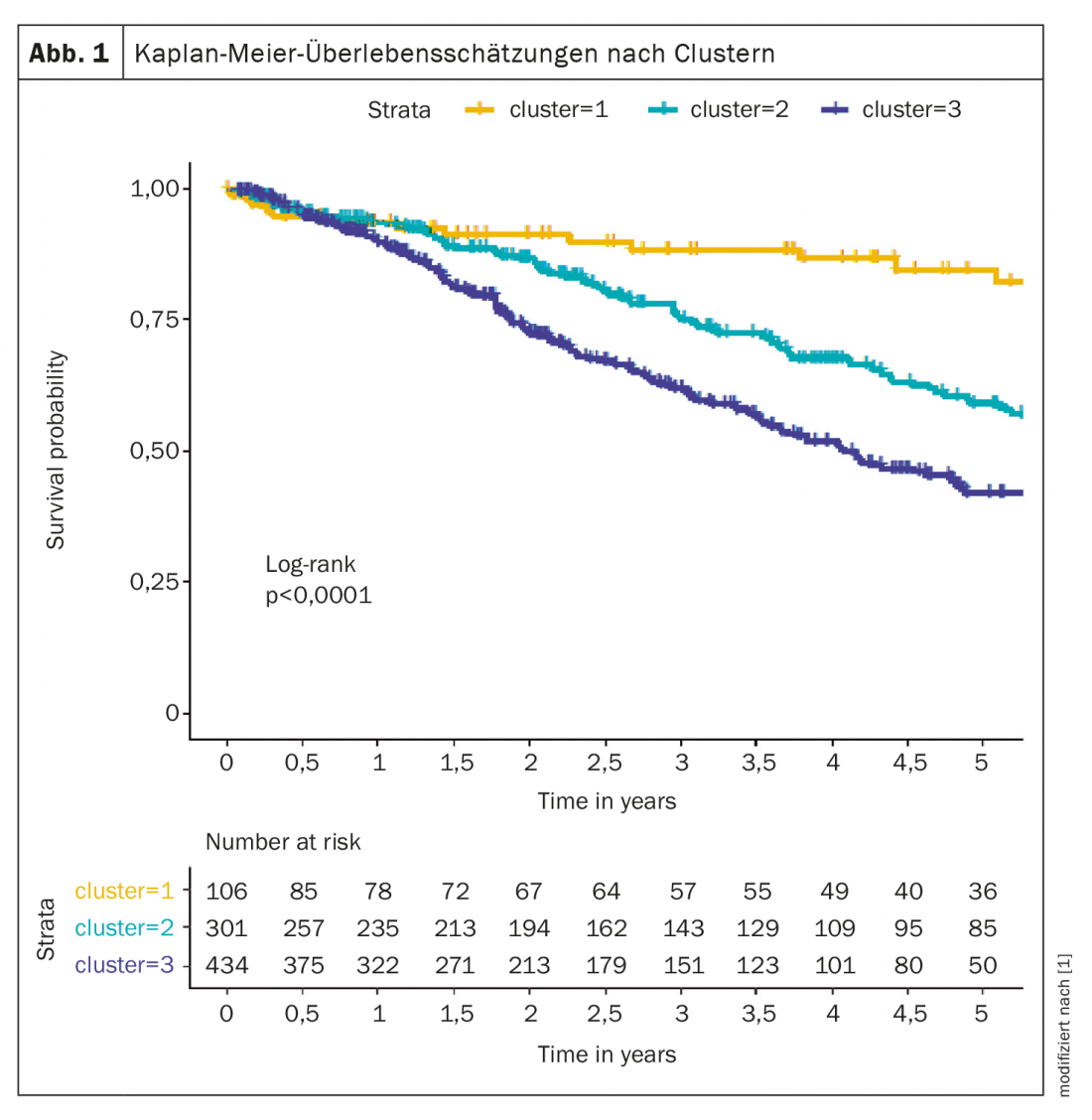
Por ejemplo, un análisis de conglomerados identificó diferentes fenotipos que difieren en la presentación clínica, la respuesta a la terapia y la supervivencia. Los grupos analizados se compusieron a partir del registro COMPERA [1], se definieron tres clusters: el cluster 1 (n=106; 12,6%) incluía una edad media de 45 años, el 76% eran mujeres, sin comorbilidades, en su mayoría nunca fumadores, DLCO ≥45%; el cluster 2 (n=301; 35,8%) incluía pacientes cuya edad media era de 75 años, el 98% mujeres, comorbilidades frecuentes, sin antecedentes de tabaquismo, DLCO en su mayoría ≥45%; el cluster 3 (n=434; 51,6%) constaba de sujetos con una edad media de 72 años, el 72% eran hombres, comorbilidades frecuentes, antecedentes de tabaquismo y DLCO en su mayoría inferior al 45%.
Los pacientes del grupo 1 respondieron mejor al tratamiento de la HAP que los pacientes de los otros dos grupos. La tasa de supervivencia a cinco años fue del 84,6% en el grupo 1, del 59,2% en el grupo 2 y del 42,2% en el grupo 3 (Fig. 1). Según el experto, estos datos sugieren que se necesitan criterios para distinguir a los pacientes con HAPI atípica de los verdaderos pacientes con HAPI.
Diagnóstico diferencial a veces difícil
Dado que las consecuencias terapéuticas dependen en gran medida de la causa de la hipertensión pulmonar, es importante completar los procedimientos diagnósticos y determinar la causa principal de la HP antes de tomar una decisión sobre la medicación para la HAP. El Simposio Mundial sobre Hipertensión Pulmonar (WSPH) ha elaborado unas directrices para tomar estas importantes decisiones. La HP del grupo 2 o las enfermedades complejas del desarrollo con aumento de la presión poscapilar pueden reconocerse con relativa facilidad por el aumento de las presiones arteriales pulmonares de enclavamiento. La HP del grupo 4 puede detectarse o excluirse mediante gammagrafías pulmonares de perfusión en combinación con un TAC torácico. La HAP del grupo 1 y la HP del grupo 3 son perfiles de enfermedad bastante diferentes, pero a veces pueden resultar difíciles de distinguir. La WSPH sugiere que la hipertensión pulmonar grave combinada con un deterioro leve en la prueba de función pulmonar (FEV1 >60 y FVC >60%), anomalías parenquimatosas leves en la TC de alta resolución del tórax y deterioro circulatorio en la prueba de ejercicio cardiopulmonar son indicativos de HAP de grupo 1. Estos pacientes son candidatos a la terapia de la HAP. Si el paciente padece una HP del grupo 3, la única indicación posible para el tratamiento de la HAP es la hipertensión pulmonar grave (mPAP ≥35 mmHg o mPAP entre 25 y 35 mmHg junto con un índice cardiaco (IC) muy bajo <2,0 L/min/m2), que sólo puede derivarse de forma invasiva, señaló el profesor Olschewski.
Un estudio también analizó la diferenciación entre pacientes con HAP (grupo 1) y pacientes con insuficiencia cardiaca (grupo 2) [2]. Aunque el aumento de las presiones de llenado del lado izquierdo y la regurgitación mitral funcional son las causas principales de la HP poscapilar, las directrices y recomendaciones diferencian entre la HP poscapilar aislada (HPpc) y la HP poscapilar y precapilar combinada (HPpc). Esta última se define por una resistencia vascular pulmonar (RVP) aumentada a unidades Wood (WE). Es importante diferenciar entre la definición general de HP (mPAP >20 mmHg) y la definición de HP precapilar que incluye la HAP, para la que también se requiere una presión arterial pulmonar en cuña (PAWP) ≤15 mmHg y un aumento de la resistencia vascular pulmonar (PVR) a ≥3 unidades Wood. Según el estudio, el tratamiento farmacológico dirigido para la HAP está indicado para una PAWP ≤15 mmHg.
Tampoco es fácil diferenciar a los pacientes con HAP y EPOC (grupo 1) de los pacientes con HP debida a EPOC (grupo 3). La mayoría de los pacientes con EPOC con HP pertenecen al grupo 3. Algunos pacientes con EPOC con HP y aumento de la presión de llenado del ventrículo izquierdo (HP poscapilar) causada por una enfermedad cardiovascular concomitante se asignan al grupo 2. Las enfermedades tromboembólicas crónicas también pueden causar HP, especialmente porque la EPOC es un factor de riesgo de tromboembolismo venoso, estos pacientes con EPOC suelen asignarse al grupo 4. Se cree que los pacientes con una obstrucción muy leve de las vías respiratorias periféricas y una HP precapilar grave con una resistencia vascular pulmonar (RVP) muy aumentada y un gasto cardíaco (GC) bajo padecen predominantemente HAP (grupo 1) con EPOC leve como afección asociada. En la mayoría de los casos, la HP es relativamente leve en los pacientes con EPOC, explicó el neumólogo, pero en un subconjunto de pacientes con EPOC, la presencia de ciertas características clínicas sugiere un “fenotipo vascular pulmonar”. Dicho fenotipo se caracterizaría por una HP precapilar grave con una resistencia vascular pulmonar notablemente aumentada, una limitación moderada del flujo aéreo, una capacidad de difusión del monóxido de carbono muy reducida, normo o hipocapnia, una limitación de la carga circulatoria y una insuficiencia cardiaca derecha progresiva.
Otro estudio intentó determinar umbrales hemodinámicos relevantes desde el punto de vista pronóstico para la HP grave en la EPOC utilizando un enfoque no sesgado [3]. Se descubrió que una RVP >5 WU era el factor hemodinámico independiente más potente para predecir la mortalidad en pacientes con EPOC. Este umbral es el mejor para identificar a los pacientes de EPOC con enfermedad vascular pulmonar grave.
Avances en la terapia farmacológica
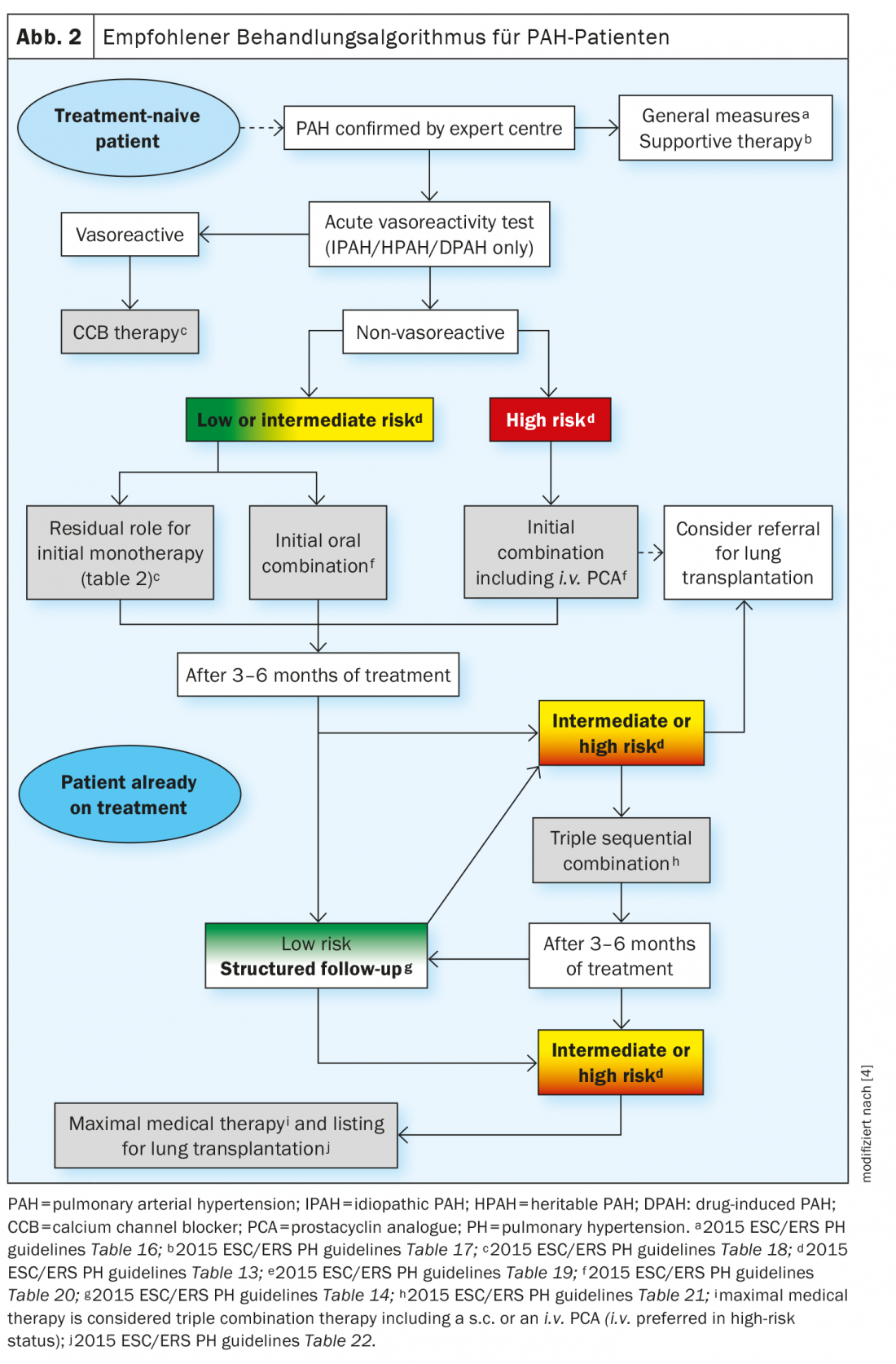
Los recientes avances en el tratamiento farmacológico de la HAP no se deben al descubrimiento de nuevas vías de señalización, sino al desarrollo de nuevas estrategias para el tratamiento combinado y a la intensificación de los tratamientos basada en la evaluación sistemática de la respuesta clínica. La estrategia de tratamiento se basa en la gravedad del paciente con HAP recién diagnosticada, que se determina mediante un enfoque multiparamétrico de estratificación del riesgo. Los parámetros clínicos, de ejercicio, de función ventricular derecha y hemodinámicos se combinan para definir el estado de riesgo bajo, intermedio o alto en función de la mortalidad esperada a 1 año. El algoritmo de tratamiento actual proporciona la estrategia inicial más adecuada, incluyendo monoterapia, terapia combinada dual o triple. Si no se alcanza el estado de bajo riesgo en las visitas de seguimiento programadas, será necesario seguir intensificando el tratamiento. En los casos más avanzados, puede ser necesario un trasplante de pulmón con la máxima terapia médica (Fig. 2).
Entre los medicamentos aprobados para el tratamiento oral se encuentran los inhibidores de la fosfodiesterasa 5 (PDE5i) como el sildenafilo y el tadalafilo, los antagonistas de los receptores de endotelina (ARE) como el bosentán, el armbrisentán y el macitentán, los estimuladores de la guanilato ciclasa soluble (sGC) como el riociguat y los agonistas de los receptores de prostaciclina como el selexipag. También es posible tratar la HAP con terapia inhalada o infusiones continuas.
Estudios prospectivos sobre la terapia
Por ejemplo, el riociguat y los inhibidores de la fosfodiesterasa-5 (PDE5i), aprobados para el tratamiento de la hipertensión arterial pulmonar (HAP), actúan a través de mecanismos diferentes en la misma vía. Por ello, el ensayo REPLACE investigó si el riociguat podría ser una opción alternativa para los pacientes con HAP que no responden adecuadamente al tratamiento con PDE5i. El objetivo era evaluar los efectos del cambio a riociguat desde el tratamiento con PDE5i en comparación con el tratamiento continuado con PDE5i en pacientes con HAP con riesgo intermedio de mortalidad a 1 año. Los resultados muestran que pasar del tratamiento con PDE5i al riociguat, que actúan ambos a través de la vía de señalización entre la guanilato ciclasa soluble en óxido nítrico y el monofosfato de guanosina cíclico, puede ser una opción estratégica para la intensificación del tratamiento en pacientes con HAP con un riesgo intermedio de mortalidad a 1 año [5].
Sotatercept, un nuevo antagonista de la activina, se une a las activinas y a los factores de diferenciación del crecimiento en un intento de restablecer el equilibrio entre las vías de señalización que promueven el crecimiento y las que lo inhiben. En el ensayo PULSAR, 24 semanas de tratamiento con sotatercept en pacientes con hipertensión arterial pulmonar que recibían tratamiento de fondo para la misma dieron lugar a una reducción de la resistencia vascular pulmonar [6]. El rendimiento físico (medido por la distancia de 6 minutos andando) y los niveles de NT-proBNP también mejoraron con el sotatercept, según el Prof. Olschewski.
“Sensación” de EE.UU.
El Prof. Olschewski tenía una “sensación” que comunicar desde EE.UU: Nunca antes se había aprobado una terapia para la HP en enfermedades pulmonares (grupo 3). Eso ha cambiado ahora, al menos en Estados Unidos. El treprostinil inhalado se utilizó allí por primera vez en este grupo de pacientes. En el ensayo INCREASE, controlado con placebo y de 16 semanas de duración, participaron enfermos con enfermedad pulmonar intersticial e hipertensión pulmonar a los que se administró treprostinil inhalado mediante un nebulizador de ultrasonidos con administración pulsátil en hasta 12 inhalaciones (72 μg en total) cuatro veces al día. En comparación con el placebo, el treprostinil inhalado mejoró significativamente el rendimiento físico de los pacientes medido por la prueba de marcha de 6 minutos [7]. En EE.UU., el principio activo ya ha sido aprobado por la FDA, pero aún está abierto si el fabricante también lo solicitará a la Agencia Europea del Medicamento (EMA) en un futuro próximo o si esperará a realizar más estudios, según explicó el experto. “En cualquier caso, es un rayo de esperanza”.

Se establece con éxito un entrenamiento de movimiento estandarizado
En el ensayo controlado aleatorizado EU-TRAIN de entrenamiento con ejercicios en pacientes con hipertensión arterial pulmonar (HAP) e hipertensión pulmonar tromboembólica crónica (HPTEC), realizado en 11 centros de 10 países europeos en una amplia población de pacientes, se observó una mejora significativa y clínicamente significativa en el criterio de valoración primario 6MGT y en los criterios de valoración secundarios WHO-FC, CdV y consumo máximo de oxígeno [8]. El estudio demostró por primera vez que un programa de ejercicio seguro y eficaz como complemento a la terapia farmacológica puede estandarizarse y aplicarse en distintos países con sistemas sanitarios diferentes.
Fuente: Pneumo Update 2021: Hipertensión pulmonar, 12.11.2021
Literatura:
- Hoeper, et al: Fenotipos de hipertensión arterial pulmonar idiopática determinados mediante análisis de conglomerados a partir del registro COMPERA. J Heart Lung Transplant 2020, doi: 10.1016/j.healun.2020.09.011.
- Rosenkranz, et al: Hipertensión pulmonar en la IC-FEp y la IC-FEr: Fisiopatología, diagnóstico, enfoques terapéuticos. Corazón 2019, doi: 10.1007/s00059-019-4831-6.
- Zeder, et al: La resistencia vascular pulmonar elevada predice la mortalidad en pacientes con EPOC. Eur Respir J 2021, doi: 10.1183/13993003.00944-2021.
- Galiè, et al: Estratificación del riesgo y terapia médica de la hipertensión arterial pulmonar. Revista Respiratoria Europea 2019, doi: 10.1183/13993003.01889-2018.
- Hoeper, et al: Cambio a riociguat frente a terapia de mantenimiento con inhibidores de la fosfodiesterasa-5 en pacientes con hipertensión arterial pulmonar (REPLACE): un ensayo multicéntrico, abierto, aleatorizado y controlado. Lancet Respir Med 2021, doi: 10.1016/S2213-2600(20)30532-4.
- Humbert, et al: Sotatercept para el tratamiento de la hipertensión arterial pulmonar. N Engl J Med 2021, doi: 10.1056/NEJMoa2024277.
- Waxman, et al: Treprostinil inhalado en la hipertensión pulmonar debida a enfermedad pulmonar intersticial. N Engl J Med 2021, doi: 10.1056/NEJMoa2008470.
- Grüning, et al: El entrenamiento con ejercicios estandarizados es factible, seguro y eficaz en la hipertensión pulmonar arterial y tromboembólica crónica: resultados de un gran ensayo controlado aleatorizado multicéntrico europeo. Eur Heart J 2021, doi: 10.1093/eurheartj/ehaa696.
InFo NEUMOLOGÍA Y ALEROLOGÍA 2022; 4(1): 20-22