Los depósitos de mucina son posibles en todas las capas de la piel, la localización del depósito no es patognomónica. Además de las mucinosis primarias, esta dermatosis por depósito se manifiesta a menudo como un epifenómeno. La etiopatogenia no se conoce del todo.
En la ZDFT de este año, el Dr. Reinhard Dummer, médico jefe de la Clínica Dermatológica del Hospital Universitario de Zúrich, y su equipo (Dr. Thierry Nordmann, MD, Dra. Carole Guillet, MD) hablaron sobre las mucinosis [1] como parte del tema principal de las dermatosis por depósito. La mucina depositada es una sustancia formada por una mezcla de glicosaminoglicanos que también se encuentran en la piel sana en forma libre (ácido hialurónico) o unida (proteoglicanos). Los glucosaminoglicanos son elementos del tejido conjuntivo que fijan el agua, desempeñan un papel importante en la consistencia y la turgencia de la piel y son producidos por los fibroblastos o los queratinocitos. Patogénicamente, se produce un aumento de la síntesis o una alteración de la degradación de los glucosaminoglicanos.
Desafío diferencial
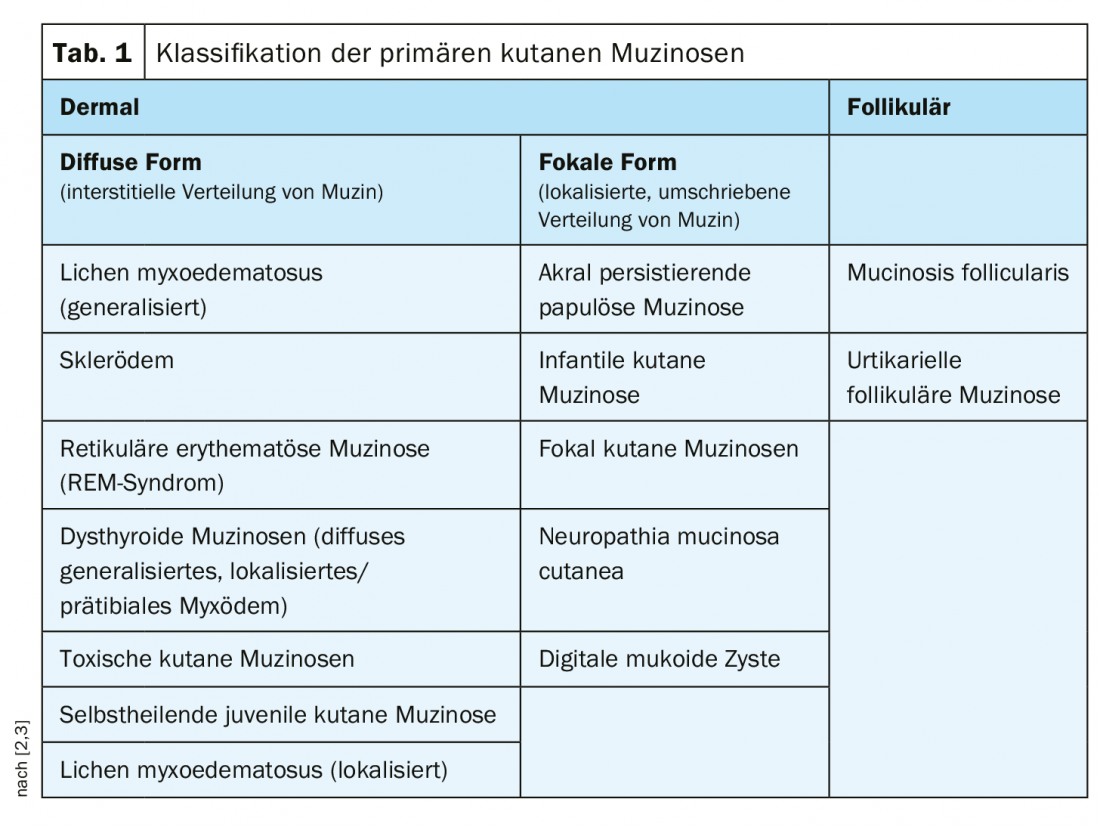
En las mucinosis cutáneas primarias, se distingue entre subtipos dérmicos y foliculares; los subtipos dérmicos se dividen en una forma difusa (distribución intersticial de la mucina) y una forma focal (distribución circunscrita localizada de la mucina) (Tab. 1) [2,3]. Las mucinosis primarias son bastante raras; más frecuente es la aparición de esta dermatosis por depósito en el contexto de procesos inflamatorios y proliferativos (fenómeno reactivo). En el lupus eritematoso y también en la dermatosis granulomatosa granuloma anular (DD necrobiosis lipoídica), una mayor presencia de mucina es un criterio de diagnóstico diferencial. Pero la proliferación de mucina también puede producirse en muchos otros procesos inflamatorios. Además, en el estroma tumoral de los tumores mesenquimales y anexiales, el tejido conjuntivo puede ser rico en mucina. En la tabla 2 se resume una clasificación de las mucinosis dérmicas como síntoma de diversas enfermedades primarias.

Síndrome REM: controversia sobre la etiopatogenia
La mucinosis eritematosa reticular, también conocida como síndrome REM, se describió por primera vez en 1974 [1,3]. La etiopatogenia del síndrome REM no se ha aclarado completamente hasta la fecha; entre otras cosas, se discute si podría tratarse de una variante clínica del lupus eritematoso cutáneo [3]. Las características clínicas típicas son placas estriadas, reticulares, urticariales, ligeramente infiltradas y enrojecidas en la zona central superior del tronco, que afectan mayoritariamente a mujeres jóvenes [3]. La exposición a la luz solar puede provocar un agravamiento de los síntomas [1]. En cuanto a la manifestación clínica, existen solapamientos entre el síndrome REM y el lupus eritematoso tumidus (forma progresiva del lupus eritematoso cutáneo) y la infiltración linfocítica de tipo Jessner y Kanof. Hasta ahora, no está claro hasta qué punto estas enfermedades representan entidades independientes [3]. Los fibroblastos de pacientes con síndrome REM muestran una respuesta desviada de la norma a la estimulación con IL-1β exógena, que desempeña un papel en el metabolismo del ácido hialurónico posiblemente desregulado en el síndrome REM [1]. Las características diagnósticas diferenciales entre el síndrome REM y el lupus tumidus se resumen en la tabla 3.

Liquen mixoedematoso: Tenga en cuenta los diagnósticos de exclusión
El liquen mixoedematoso es una mucinosis primaria poco frecuente con un curso crónico progresivo [3]. Se distinguen dos subtipos: la forma localizada y la forma generalizada (Tab. 4) [1]. Este último también se denomina escleromixoedema y se caracteriza por rasgos esclerodermiformes y papulares.
Los lugares de predilección del liquen mixoedematoso localizado son el dorso de las manos, los lados extensores de los brazos, la parte superior del tronco, la cara y las axilas [3]. Los rasgos típicos son pápulas rojizas individuales del color de la piel que pueden confluir y aparecer como liquenificación y/o engrosamiento general de la piel. La distribución de la mucina en la forma localizada puede ser difusa o focal. Existe controversia en la literatura sobre si el liquen mixoedematoso discreto, la mucinosis papular persistente acral, la mucinosis papular autocurativa (forma juvenil/adulta), la mucinosis infantil y la forma nodular deben considerarse subtipos distintos del liquen mixoedematoso localizado. Los síntomas clásicos del subtipo generalizado (escleromixoedema) son un engrosamiento eritematoso generalizado de la piel, que puede provocar una rigidez mímica y una restricción del movimiento articular; en algunos casos, se han observado anomalías cardiovasculares además de miopatías y síntomas neurológicos [3]. El diagnóstico diferencial del escleromixoedema debe diferenciarse clínica e histológicamente de otros cambios papulares como la esclerodermia sistémica y el escleroedema. Un rasgo distintivo de otras mucinosis son los hallazgos histológicos característicos en el liquen mixoedematoso local y el escleromixoedema. Se caracterizan, entre otras cosas, por una deposición pronunciada de mucina en la dermis media y superior y, a menudo, un aumento de fibroblastos con núcleos grandes en forma de estrella; además, las fibras de colágeno pueden estar compactadas y aumentadas (fibromucinosis) y puede haber un pequeño infiltrado linfocítico perivascular [3]. Como terapia para el liquen mixoedematoso se recomiendan los glucocorticoides tópicos y los inhibidores tópicos de la calcineurina; también pueden utilizarse tratamientos con PUVA y láser. En algunos casos, se consiguió reducir los síntomas con citostáticos (ciclofosfamida, clorambucil, melfalán), isotretinoína y plasmaféresis, y en otros con inmunoglobulinas intravenosas, dosis altas de melfalán y trasplante autólogo de células madre de sangre periférica [3]. El conferenciante señala que, en presencia de liquen mixoedematoso localizado, la malignidad y la enfermedad tiroidea son diagnósticos importantes que hay que excluir [1].

Mucinosis folicular: ostia folicular prominente
A diferencia del síndrome REM y el liquen mixoedematoso, que son subtipos dérmicos, la mucinosis folicular se caracteriza histológicamente por depósitos de mucina en los folículos. También hay un infiltrado linfocítico atípico y degeneración del epitelio [1]. Clínicamente, el subtipo folicular se manifiesta como pápulas y placas elevadas focales, foliculares, de color piel a eritematosas, con ostia folicular prominente (a veces con parches queratósicos similares a comedones). Etiopatogénicamente, se discute la inflamación linfocítica inicial como posible causa de la acumulación secundaria de mucina. Se cree que existe una transición continua de la mucinosis folicular (más común en pacientes jóvenes) a la micosis fungoide foliculotrópica (más común en pacientes mayores) [1].
Depósitos de mucina en el contexto de enfermedades multisistémicas
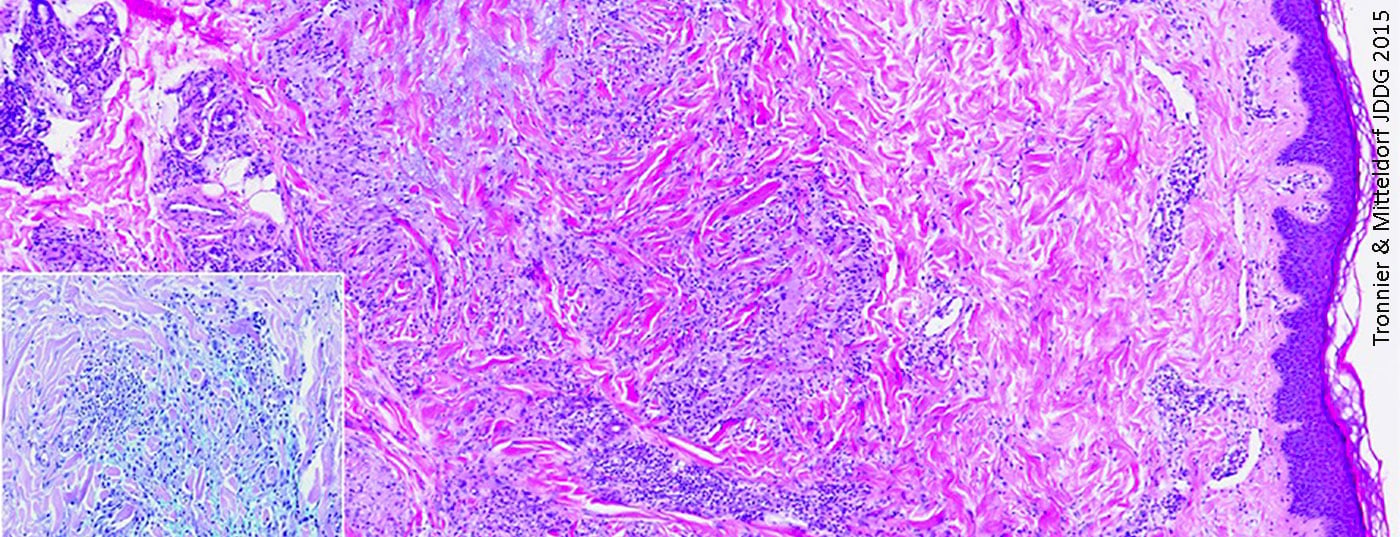
El granuloma anular (Fig. 1) es una enfermedad granulomatosa no infecciosa de etiología desconocida [6], que se presenta con frecuencia en pacientes jóvenes [7]. Los lugares de predilección de los síntomas, que suelen manifestarse en forma de placas anulares o arciformes, son principalmente las extremidades [7]. La deposición de mucina en la zona de degeneración del tejido conjuntivo es una característica histológica importante [8]. Esto es fácilmente reconocible histopatológicamente en las tinciones con azul Alcian o hierro coloidal [8]. En su mayor parte, la epidermis no está afectada; en la dermis hay tejido conectivo degenerado rodeado de células epitelioides dispuestas en forma de empalizada ligeramente alargadas [8]. Además de una forma solitaria, también existe una forma diseminada, que puede darse, por ejemplo, en el contexto de la diabetes, y que afecta principalmente a los casos de edad avanzada [7].

El lupus eritematoso es una enfermedad inflamatoria autoinmune con manifestaciones clínicas y cursos heterogéneos [9]. El lupus eritematoso sistémico es una enfermedad multisistémica potencialmente mortal con afectación cutánea. La aparición de lesiones cutáneas durante el curso de esta enfermedad es frecuente y puede darse hasta en un 70-85 % de los pacientes [9]. En el lupus eritematoso cutáneo agudo, se distingue una forma localizada (eritema en mariposa) de una variante generalizada (exantema maculopapular), que es el subtipo más común del lupus eritematoso sistémico (30-60%) [9]. Histopatológicamente, una dermatitis de interfase pobre en células y un infiltrado linfocítico perivascular más bien escaso con depósitos de mucina son hallazgos típicos del lupus eritematoso cutáneo agudo [9].
La dermatomiositis es otra enfermedad primaria con depósitos dérmicos de mucina como característica histopatológica típica [10]. Éstas se producen en el contexto de focos inflamatorios en la zona de la capa de transición dermoepidérmica [10].
Fuente: ZDFT, Zúrich
Literatura:
- Dummer R, Guillet C, Nordmann Th: Presentación de diapositivas: Tema anual – dermatosis deposicionales. Mucinosis. Prof. Reinhard Dummer y equipo, IX Jornadas de Formación Dermatológica 2019, Zúrich, 26 de junio de 2019.
- Rongioletti F, Rebora A: Mucinosis cutáneas: criterios microscópicos para el diagnóstico. Am J Dermatopathol 2001; 23: 257-267.
- Kuhn A: Mucinosis. Dermatología, venereología y alergología de Braun-Falco 2017; 1-9
https://link.springer.com/referenceworkentry/10.1007%2F978-3-662-49546-9_93-1) - Schallera J, Meigelb WN: Dermatosis deposicionales, en: Histopathology of the Skin ed: Cerroni L et al, 2ª edición 2016, Springer: Berlin Heidelberg. DOI 10.1007/978-3-662-44367-5_24-1, https://link.springer.com/content/pdf/10.1007%2F978-3-662-44367-5_24-1.pdf
- Rongioletti F, et al: Mucinosis eritematosa reticular: revisión de las características de los pacientes, afecciones asociadas, terapia y resultado en 25 casos. Br J Dermatol 2013; 169: 1207-1211.
- Piette EW, Rosenbach M: Granuloma anular. Patogénesis, asociaciones y desencadenantes de la enfermedad y opciones terapéuticas. JAAD 2016; 75 (3): 467-479. www.jaad.org/article/S0190-9622(15)01500-5/abstract
- Houriet C, et al.: Hautmanifestationen bei inneren Erkrankungen – 1.Teil, Universitätsklinik für Dermatologie, Inselspital Bern. Schweiz Med Forum 2013;13(47): 949-953, https://boris.unibe.ch/45902/1/houriet_smf_1.pdf
- Tronnier M, Mitteldorf C: Características histológicas de las enfermedades granulomatosas de la piel: Parte 1: Enfermedades granulomatosas no infecciosas. Minirevista. JDDG 2015; 13 (3): 211-216, https://onlinelibrary.wiley.com/doi/full/10.1111/ddg.12610_suppl
- Sticherling M, Kuhn A: Lupus eritematoso. Dermatología, Venereología y Alergología de Braun-Falco 2017; 1-18, https://link.springer.com/referenceworkentry/10.1007%2F978-3-662-49546-9_54-1
- Miller ML: Diagnóstico y diagnóstico diferencial de la dermatomiositis y la polimiositis en adultos. UpToDate, www.uptodate.com/contents/diagnosis-and-differential-diagnosis-of-dermatomyositis-and-polymyositis-in-adults
DERMATOLOGIE PRAXIS 2019; 29(5): 41-43 (publicado el 10.10.19, antes de impresión).









