La sobrecarga de hierro es una consecuencia inevitable y potencialmente mortal de las transfusiones múltiples de concentrados de hematíes. Como las manifestaciones clínicas son inespecíficas y suelen desarrollarse lentamente, esta complicación suele pasarse por alto.
La sobrecarga de hierro es una consecuencia inevitable y potencialmente mortal de las transfusiones múltiples de concentrados de hematíes. Como las manifestaciones clínicas son inespecíficas y suelen desarrollarse lentamente, esta complicación suele pasarse por alto.
En general, hoy en día ya no se distingue entre sobrecarga de hierro primaria y secundaria porque tal definición depende, entre otras cosas, de la sensibilidad de los métodos de examen. El término “siderosis” suele utilizarse para describir la deposición de hierro sin daño tisular, como ocurre en la deposición de hierro localizada y relacionada con hemorragias (por ejemplo, la siderosis pulmonar).
Aparición de sobrecarga de hierro
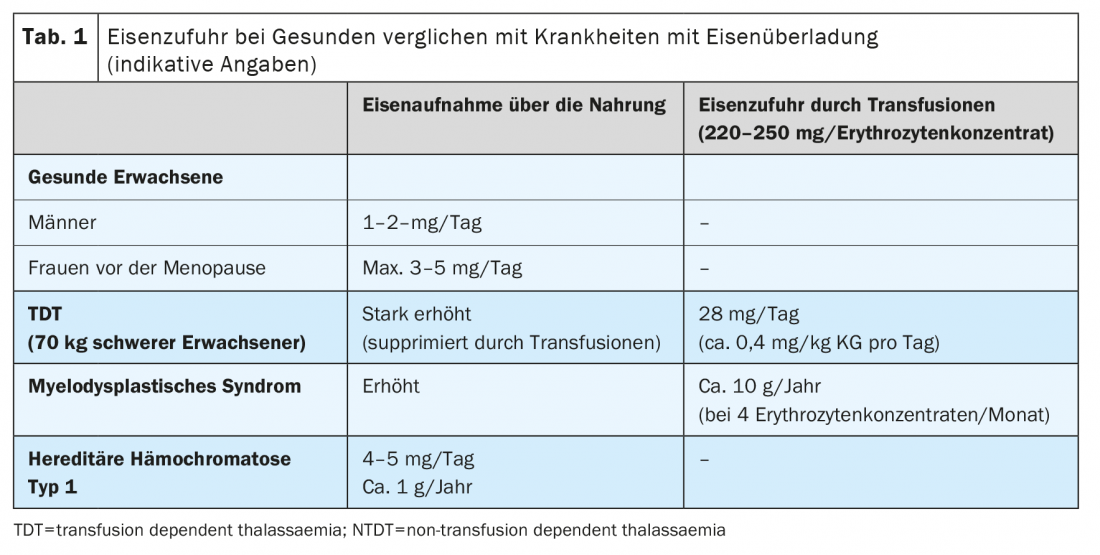
Una dieta occidental típica contiene unos 6 mg de hierro/1.000 kcal, de los cuales sólo se absorben normalmente 1-2 mg/día (alrededor del 10%) y un máximo de 3-5 mg/día cuando la absorción intestinal de hierro está aumentada. El almacenamiento normal de hierro es de 500-1000 mg en los hombres y de 300-400 mg en las mujeres premenopáusicas y se encuentra sobre todo en el hígado. El organismo no dispone de mecanismos fisiológicos activos para excretar el exceso de hierro. Los adultos sanos excretan alrededor de 1 mg de hierro/día a través de la piel y las células gastrointestinales, las mujeres premenopáusicas pierden una media adicional de 0,5-1 mg/día a través de la menstruación.
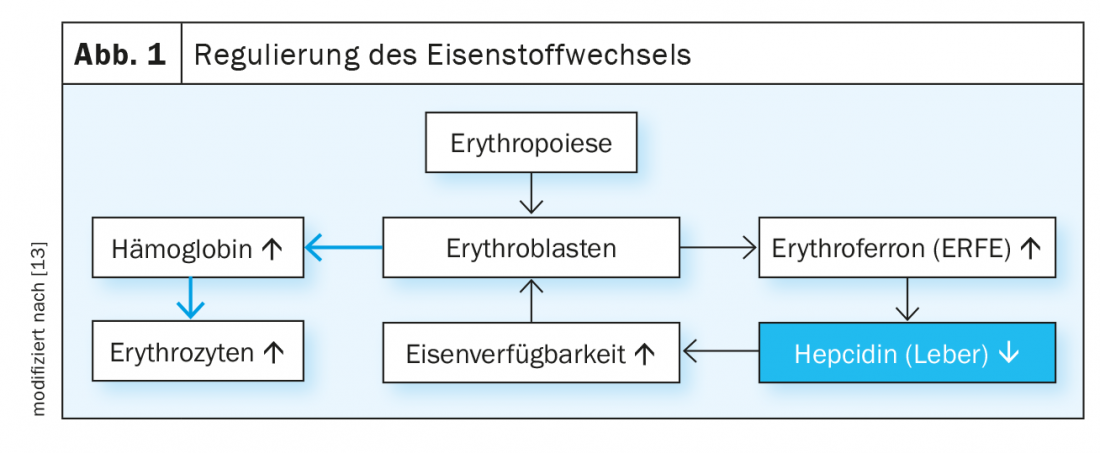
Los niveles plasmáticos de hierro están regulados por el sistema hepcidina/ferroportina (Fig. 1). La hormona peptídica hepcidina induce la degradación de la proteína exportadora de hierro ferroportina. La ferroportina se expresa principalmente en las células de la mucosa duodenal, las células hepáticas y los macrófagos y media en la regulación de la absorción de hierro de los alimentos, la liberación de hierro del hígado según sea necesario y el reciclaje de hierro en los macrófagos. Con una disponibilidad suficiente de hierro, la síntesis hepática de hepcidina aumenta, bloqueando una mayor absorción gastrointestinal de hierro. Por otro lado, la anemia crónica y la eritropoyesis ineficaz provocan la inhibición de la síntesis de hepcidina en el hígado y, por tanto, un aumento de la absorción de hierro en el duodeno. La hormona eritroferrón, que se produce bajo la influencia de la eritropoyetina en los eritroblastos, también suprime la producción de hepcidina y estimula así la absorción de hierro y la movilización a partir de las reservas en situaciones de estrés eritropoyético.
El almacenamiento excesivo de hierro se debe a dos mecanismos principales: El aporte iatrogénico de hierro a través de transfusiones de concentrados de glóbulos rojos y el aumento de la ingesta de hierro a través de los alimentos. Cada concentrado de hematíes contiene 220-250 mg de hierro. En pacientes adultos, la deposición relevante de hierro se produce después de 15-20 transfusiones, en niños pequeños ya después de más de 10 administraciones de concentrados de glóbulos rojos. El aumento de la absorción gastrointestinal desempeña un papel central en todas las situaciones de eritropoyesis aumentada o ineficaz. En este caso, los eritroblastos perecen por muerte celular en la médula ósea antes de que puedan madurar y convertirse en eritrocitos. Esto conduce a una sobreestimulación e hiperplasia de la eritropoyesis, que varía en función de la enfermedad subyacente. Cuanto más pronunciada es la alteración de la eritropoyesis, más se incrementa la absorción de hierro de los alimentos (Tab. 1).
En la hemocromatosis hereditaria, un trastorno genético del metabolismo del hierro sin anemia, la deposición de hierro es el resultado de la desregulación del eje hepcidina-ferroportina, más comúnmente por la disminución de la producción de hepcidina (como en la hemocromatosis hereditaria de tipo 1). Más raramente, existe una disfunción de otras moléculas reguladoras del hierro.
Dependiendo de la enfermedad, el curso temporal y el alcance de la acumulación de hierro, así como su distribución en los órganos, difieren. También influyen otros factores como las infecciones crónicas (por ejemplo, la hepatitis crónica) y las enfermedades metabólicas (por ejemplo, la esteatohepatitis).
Efectos del exceso de hierro en las células
La toxicidad asociada al hierro se observa principalmente en los tejidos que almacenan hierro en altas concentraciones. Se trata, en particular, del hígado, el sistema endocrino y el miocardio. El hierro de las transfusiones se deposita primero en los macrófagos del hígado, el bazo y la médula ósea, después en los hepatocitos y sólo más tarde en los órganos endocrinos (sobre todo en el páncreas) y en el músculo cardiaco.
El hierro circulante está ligado a la proteína transferrina y luego es entregado a las células por la unión de la transferrina a los receptores apropiados (TfR1, expresado en todas las células, y TfR2, presente en los hepatocitos). La proporción normal de transferrina unida al hierro es del 16-45% y se mide como saturación de transferrina. En presencia de un exceso de hierro, se supera la capacidad de unión de la transferrina y, a niveles superiores al 70-75%, aparecen moléculas circulantes de hierro no unido (hierro no unido a la transferrina , NTBI) que se absorben rápidamente en las células a través de vías no reguladas (por ejemplo, a través de los canales de calcio en las células del miocardio). La ingesta continuada de NTBI aumenta tanto las reservas fisiológicas de hierro intracelular (ferritina) como la aparición de formas lábiles de hierro. Estos últimos catalizan reacciones bioquímicas como la reacción de Fenton, produciendo radicales de oxígeno reactivos (ROS). Las consecuencias son la peroxidación de las moléculas intracelulares, especialmente los lípidos, con daños en los orgánulos, la muerte celular, la estimulación de la fibrogénesis y, finalmente, la disfunción de los órganos. Además, el ADN resulta dañado, lo que puede provocar la inestabilidad del genoma y una tendencia a la mutagénesis (Fig. 2) .
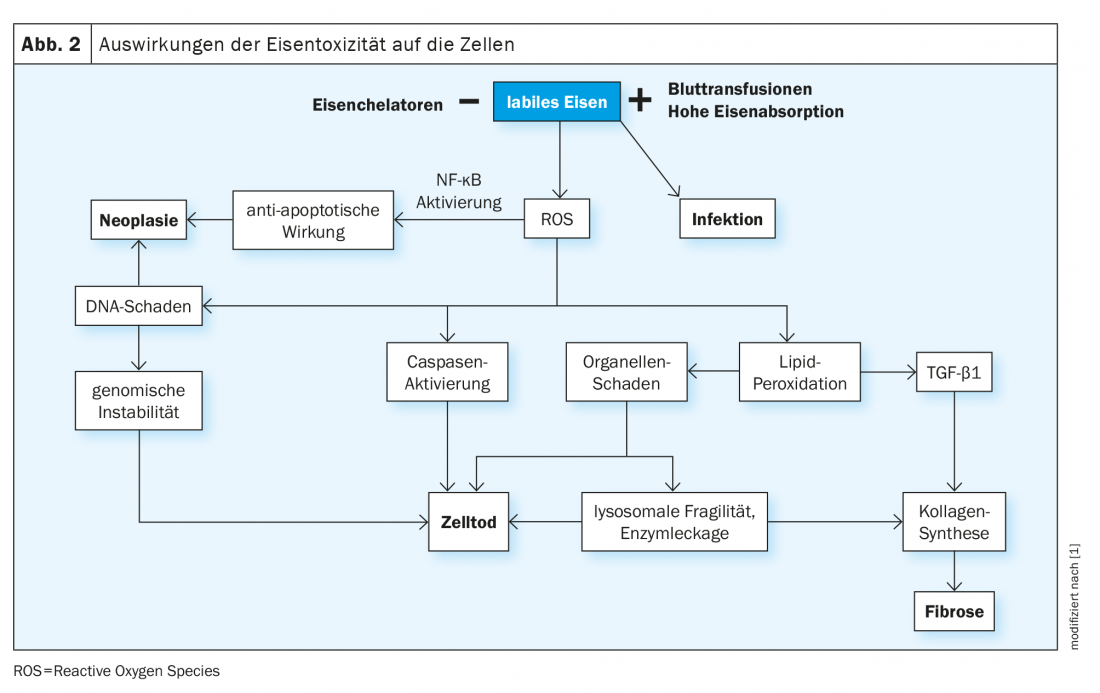
La deposición de hierro es tolerada de forma diferente por los distintos tejidos. Por ejemplo, el hígado puede almacenar mucho más hierro que el miocardio sin consecuencias perjudiciales. Otro factor decisivo para el desarrollo de daños en los órganos es la duración de la exposición de las células al NTBI. Actualmente puede persistir durante varias décadas en las enfermedades congénitas dependientes de la transfusión, como la talasemia mayor, y se asocia a un mayor riesgo de neoplasias malignas.
Un efecto subestimado de la sobrecarga de hierro es el favorecimiento del crecimiento bacteriano y el correspondiente riesgo de infecciones. La rápida disponibilidad de hierro libre para los microorganismos, por un lado, y los efectos sobre la función de los macrófagos y los leucocitos, por otro, explican la mayor susceptibilidad a las infecciones de los pacientes con talasemia dependiente de transfusiones. Las nuevas observaciones también muestran que el exceso de hierro provoca un estrés oxidativo en el endotelio de todos los vasos y una reducción de la exportación de colesterol por parte de los macrófagos de la pared vascular, lo que puede conducir a un aumento de la formación de placas.
Pacientes con riesgo de sobrecarga de hierro
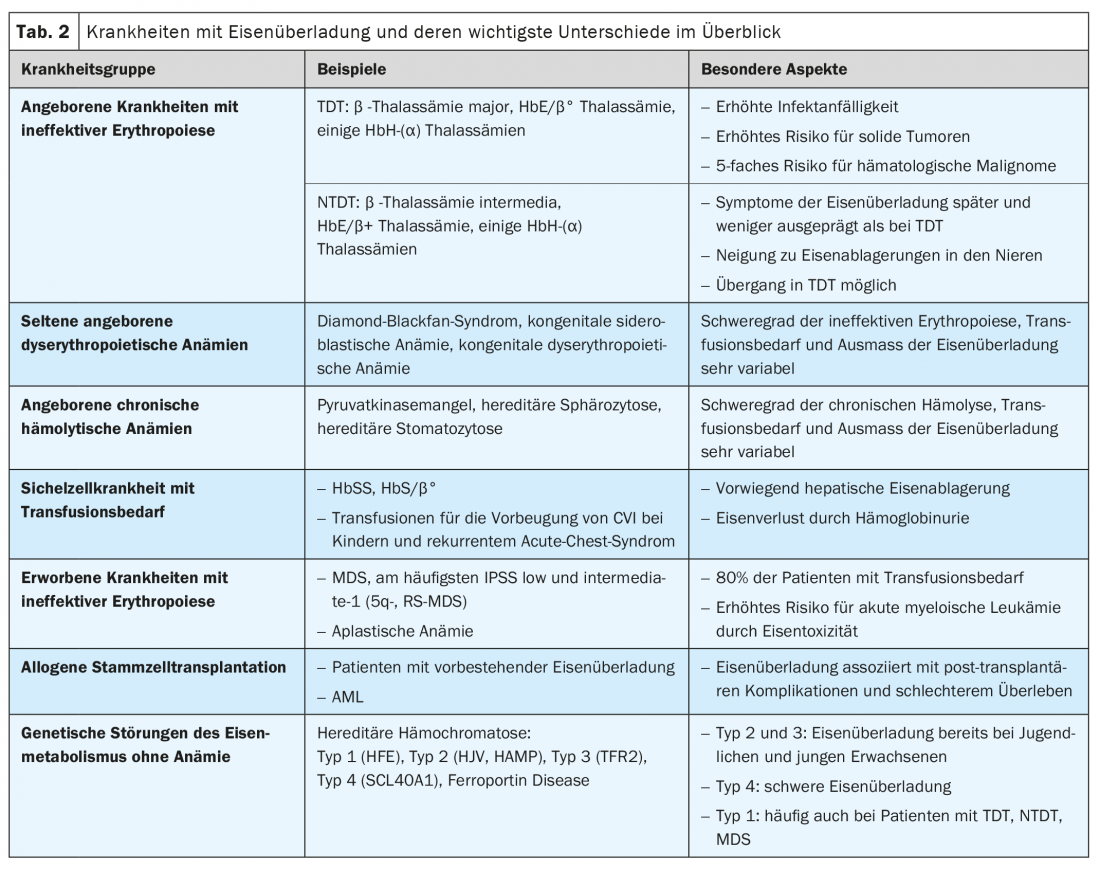
Las situaciones clínicas asociadas a la sobrecarga de hierro incluyen enfermedades congénitas y adquiridas con eritropoyesis ineficaz (por ejemplo, talasemia y síndrome mielodisplásico), enfermedades hemolíticas crónicas, anemia falciforme (ECF) que requiere transfusión y trastornos genéticos del metabolismo del hierro sin anemia (hemocromatosis hereditaria). (Tab. 2). Además, el exceso de hierro tiene importancia pronóstica en pacientes tras un trasplante alogénico de células madre.
Talasemia dependiente de transfusión (TDT) y no dependiente de transfusión (NTDT)
Los síndromes talasémicos se encuentran entre las enfermedades genéticas más comunes en todo el mundo y están causados por defectos genéticos en el cromosoma 11(β-talasemia) o 16(α-talasemia), que conducen a una síntesis reducida o ausente de las cadenas de globina correspondientes. El enorme número de variantes genéticas y sus combinaciones descritas hasta ahora, así como el tipo de herencia, explican la gran variabilidad clínica. Desde un punto de vista clínico, se distingue entre talasemias dependientes de transfusión (TDT) y no dependientes de transfusión (NTDT). Esta subdivisión corresponde aproximadamente a la de Thalassämia major e intermedia.
La β-talasemia mayor se considera el paradigma de la TDT y de las enfermedades por sobrecarga de hierro. La característica principal de la TDT es una eritropoyesis gravemente alterada o ausente, que resulta de una síntesis reducida de hemoglobina y de la precipitación de un exceso de cadenas de globina(α o β). Existe una anemia grave con, por un lado, una necesidad de transfusiones de por vida desde el primer año de vida y, por otro, una absorción intestinal de hierro estimulada al máximo. Así pues, la ingesta masiva diaria de hierro procedente de los alimentos y la iatrogénica, que por sí sola asciende a unos 0,4 mg/kg de peso corporal al día, se suman. Los síntomas clásicos de la sobrecarga de hierro en la TDT son la hepatopatía (fibrosis, cirrosis, carcinoma hepatocelular), la cardiopatía (especialmente la cardiopatía ritmogénica y dilatada), las endocrinopatías (diabetes mellitus, hipotiroidismo, hipogonadismo) y la osteoporosis (Fig. 3) .
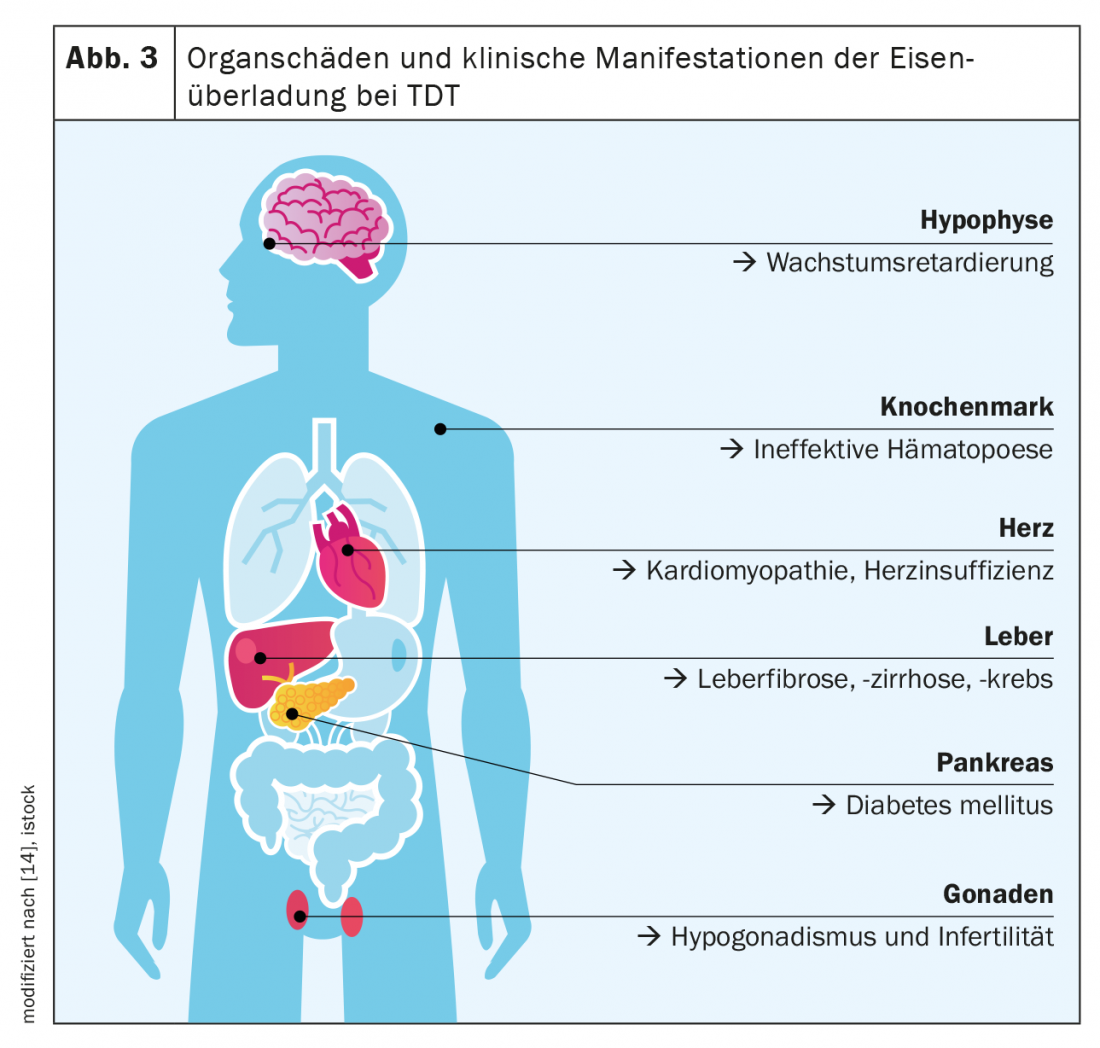
Con la introducción de la terapia transfusional en los años 70, la esperanza de vida de los pacientes con TDT se alargó hasta la pubertad, siendo el fallo cardiaco y endocrino las causas más comunes de muerte. Gracias a la posibilidad de la quelación del hierro, los pacientes con TDT tienen ahora una esperanza de vida de 40-50 años o más y presentan formas menos graves de cardiopatías y endocrinopatías. Sin embargo, existe un mayor riesgo de neoplasias malignas, especialmente carcinomas gastrointestinales, y un riesgo más de 5 veces mayor de neoplasias hematológicas como consecuencia a largo plazo de la toxicidad del hierro.
En la NTDT, el aumento de la absorción de hierro (aprox. 0,01 mg/kg de peso corporal al día) es consecuencia de un gran aumento de la eritropoyesis y representa el principal mecanismo de deposición de hierro. Dado que la acumulación de hierro en el NTDT se desarrolla lentamente, las manifestaciones clínicas no se producen hasta la infancia tardía o incluso hasta la edad adulta temprana. Los síntomas de sobrecarga de hierro suelen ser menos pronunciados, la afectación cardiaca menos frecuente y el riesgo de neoplasia menor que con la TDT. Sin embargo, los pacientes con NTDT tienen tendencia a acumular hierro en los riñones, lo que puede provocar una disfunción intersticial y glomerular. Es importante señalar que la NTDT puede convertirse en TDT a lo largo de la vida.
Anemias congénitas raras con eritropoyesis ineficaz y anemias hemolíticas crónicas congénitas
También en las anemias congénitas, como el síndrome de Diamond-Blackfan, la gravedad de la eritropoyesis ineficaz y la anemia, así como la necesidad de transfusión, determinan el grado de sobrecarga de hierro. Los datos específicos sobre estas enfermedades raras son escasos. En función de la necesidad de transfusión, que puede ser relevante incluso en niños pequeños, los riesgos y las manifestaciones clínicas de la sobrecarga de hierro son similares a los de la TDT y la NTDT.
Las enzimopatías eritrocíticas congénitas como la deficiencia de piruvato quinasa, las membranopatías (por ejemplo, la esferocitosis hereditaria) y los trastornos de la síntesis del hemo (por ejemplo, las porfirias) conducen a una hemólisis crónica más o menos pronunciada y, por tanto, a una hiperplasia de la eritropoyesis. La sobrecarga de hierro puede producirse en los individuos afectados incluso sin transfusiones (hasta en el 47% de los pacientes con deficiencia de piruvato cinasa), y los síntomas correspondientes suelen aparecer en la edad adulta y sólo en raras ocasiones en la infancia. En la esferocitosis hereditaria, que es la causa más común de hemólisis crónica con una prevalencia estimada de 1:2000 a 1:2500, sólo una proporción de pacientes requiere transfusiones esporádicas o regulares.
Enfermedad de células falciformes (ECF) con necesidad de transfusión
Las transfusiones regulares están indicadas en los pacientes con ECF sólo en situaciones específicas, como la prevención de insultos cerebrovasculares en niños y el síndrome torácico agudo recurrente o las crisis vaso-oclusivas graves. En estos casos, suelen realizarse exanguinotransfusiones manuales o automáticas, que no se asocian a un mayor aporte de hierro relacionado con la transfusión.
Los pacientes con ECF suelen perder hierro a través de la hemoglobinuria (en cantidades equivalentes a hasta 10 concentrados de hematíes al año) y presentan niveles plasmáticos bajos de NTBI. Por lo tanto, la sobrecarga de hierro sólo se desarrolla en una proporción relativamente pequeña de los afectados y los síntomas -especialmente la hepatopatía y la fibrosis hepática- aparecen clásicamente más tarde en la vida. Aunque el daño endocrino o cardiaco es poco frecuente en pacientes con MSC, las manifestaciones clínicas de la sobrecarga de hierro y la propia MSC son a menudo difíciles de distinguir, y el daño orgánico relacionado con el hierro se subestima en ocasiones. Los estudios demuestran que hasta un 11% de los pacientes con ECF mueren como consecuencia de una sobrecarga de hierro. Debido a la esperanza de vida cada vez más larga, es de esperar que las complicaciones a largo plazo asociadas al hierro también aumenten en la ECF, al igual que en la TDT y la NTDT.
Anemia crónica adquirida por insuficiencia de la médula ósea
El síndrome mielodisplásico (SMD) comprende un grupo de enfermedades adquiridas caracterizadas por un fallo de la médula ósea de diversos grados y la posibilidad de desarrollar una leucemia mieloide aguda. El SMD es una de las neoplasias hematológicas más frecuentes (incidencia de aproximadamente 4/100.000 en Europa) y afecta sobre todo a pacientes de edad avanzada (edad media en el momento del diagnóstico: 71 años).
En los SMD de bajo riesgo (IPSS bajo e intermedio-1), predominan las citopenias, con anemia debida a una eritropoyesis ineficaz en cerca del 80% de los casos. Como la mayoría de los pacientes con SMD se vuelven dependientes de las transfusiones, a menudo desarrollan una sobrecarga de hierro. Las diferencias más importantes con la TDT y la NTDT son la edad mucho mayor de los pacientes en el momento del diagnóstico y la elevada estabilidad genómica de los clones displásicos en la médula ósea. Al igual que en la TDT, las complicaciones cardiacas son frecuentes en los pacientes con SMD transfundidos regularmente (82,4% frente al 67,1% en los pacientes no transfundidos). Una importancia particular de la toxicidad del hierro en los SMD radica en el fomento de la progresión a leucemia aguda y en la influencia negativa sobre la supervivencia tras el trasplante alogénico de células madre.
Trasplante alogénico de células madre
El trasplante alogénico de células madre es un tratamiento potencialmente curativo para los pacientes con TDT, SCD y MDS y otras enfermedades con depósito de hierro. La influencia de la sobrecarga de hierro en el éxito del trasplante alogénico de células madre, especialmente en la mortalidad asociada a la terapia, se describió por primera vez en las talasemias y ahora se reconoce cada vez más en otros cuadros clínicos.
Incluso la toxicidad de la quimioterapia puede atribuirse en parte a los efectos negativos de la sobrecarga de hierro, ya que durante el acondicionamiento se produce una movilización masiva de las reservas de hierro de la médula ósea con liberación de NTBI. Las necesidades de transfusión antes y después del trasplante influyen en la evolución peritrasplante. Los datos muestran que el 88% de los pacientes con SMD y el 97% de los pacientes con leucemia mieloide aguda presentan niveles elevados de ferritina antes del trasplante y que el exceso de hierro se asocia a mucositis, obstrucción de los vasos sinusoidales hepáticos, sepsis y, en general, a una menor supervivencia global.
Trastornos genéticos del metabolismo del hierro sin anemia
El aumento de la absorción intestinal de hierro es el correlato fisiopatológico de las diversas formas de hemocromatosis hereditaria. La ingesta de hierro puede aumentarse hasta 4 mg diarios, es decir, de 2 a 4 veces más que en los individuos sin hemocromatosis hereditaria (Tabla 1) .
En la hemocromatosis hereditaria asociada al HFE (tipo 1), una de las enfermedades genéticas más comunes en la población europea, el hierro se deposita muy lentamente (alrededor de 1 g/año) y los síntomas suelen aparecer en la edad adulta (entre los 40 y los 50 años en los hombres, a menudo después de la menopausia en las mujeres). Las manifestaciones clínicas más comunes son la hepatopatía, las artropatías y la diabetes mellitus. El momento y la gravedad de la acumulación de hierro difieren en función de la mutación subyacente. Algunas formas raras como la hemocromatosis juvenil (tipo 2) son más agresivas y conducen a un almacenamiento relevante de hierro ya en la pubertad.
Debido a su frecuencia, la hemocromatosis hereditaria, especialmente la de tipo 1, se detecta con no poca frecuencia en pacientes con talasemia, SMD o riesgos de acumulación patológica de hierro, por lo que representa un factor adicional en el desarrollo de una sobrecarga de hierro potencialmente grave.
Mensajes para llevarse a casa
- El aumento de la absorción intestinal de hierro y el aporte iatrogénico de hierro a través de transfusiones de concentrados de glóbulos rojos son los principales mecanismos de desarrollo de la sobrecarga de hierro.
- La acumulación patológica de hierro se produce en enfermedades hematológicas congénitas y adquiridas con eritropoyesis ineficaz, incluso en ausencia de transfusiones regulares.
- La sobrecarga de hierro se desarrolla más rápidamente y en mayor medida en los pacientes transfundidos regularmente que en las enfermedades no dependientes de la transfusión; en los trastornos genéticos del metabolismo del hierro (hemocromatosis hereditaria), el hierro se deposita mucho más lentamente en comparación.
- En los SMD, el exceso de hierro celular puede aumentar la inestabilidad del genoma en los clones celulares preleucémicos, favoreciendo la transformación en leucemia aguda.
- La sobrecarga de hierro se asocia a complicaciones clínicas y a una mayor mortalidad tras el trasplante alogénico de células madre.
- La hemocromatosis hereditaria, debido a su frecuencia, puede estar presente en pacientes con otros riesgos de sobrecarga de hierro clínicamente relevantes y puede influir en la evolución clínica.
Literatura:
- Porter JB, et al: Nuevos conocimientos sobre la toxicidad del hierro relacionada con las transfusiones: implicaciones para el oncólogo. Crit Rev Oncología/Hematología 2016; 99: 261-271
- Camaschella C, Nai A, Silvestri L: Metabolismo del hierro y trastornos del hierro revisitados en la era de la hepcidina. Haematologica 2020; 105: 260-72
- Porter JB, Garbowski M: La fisiopatología de la sobrecarga transfusional de hierro. Hematol. Oncol Clin N Am 2014; 28: 683-701.
- Hahalis G, et al.: Disfunción vasomotora global y envejecimiento vascular acelerado en la beta-talasemia mayor. Aterosclerosis 2008; 198 (2): 448-457.
- Gardenghi S, et al.: La eritropoyesis ineficaz en la beta-talasemia se caracteriza por una mayor absorción de hierro mediada por la regulación a la baja de la hepcidina y la regulación al alza de la ferroportina. Sangre 2007; 109(11): 5027-5035.
- Vento S, Cainelli F, Cesario F: Infecciones y talasemia. Lancet Infect Dis 2006; 6(4): 226-233.
- Porter J, Garbowski M: Consecuencias y gestión de la sobrecarga de hierro en la anemia falciforme. Hematología Am Soc Hematol Educ Program. 2013; 2013: 447-456.
- Roggero S, et al: Sobrecarga grave de hierro en la anemia de Blackfan-Diamond: un estudio de casos y controles. Am J Hematol. 2009; 84: 729-32.
- Zanella S, Garani MC, Borgna-Pignatti C: Malignidades y talasemia: una revisión de la literatura. Ann N Y Acad Sci 2016; 1368(1): 140-148.
- Gattermann N: Sobrecarga de hierro en los síndromes mielodisplásicos (SMD). Int J Hematol 2018; 107: 55-63.
- Koreth J, Antin JH: Sobrecarga de hierro en neoplasias hematológicas y resultado del trasplante alogénico de células madre hematopoyéticas. Haematologica 2010; 95: 364-366.
- Pilling LC, et al: Afecciones comunes asociadas a las variantes genéticas de la hemocromatosis hereditaria: estudio de cohortes en el Biobanco del Reino Unido. BMJ 2019; 364: k5222.
- Leuenberger N, et al.: La hepcidina como biomarcador potencial del dopaje sanguíneo. Prueba de drogas Anal 2017; 9(7): 1093-1097.
- Novartis: Vivir con transfusiones. www.leben-mit-transfusionen.de/eisenueberladung/krankheitsbild (fecha de acceso: 10.02.2022)
InFo ONCOLOGÍA Y HEMATOLOGÍA 2022; 10(1): 12-17