La presencia de EPI en pacientes con artritis reumatoide limita el pronóstico, la supervivencia y la calidad de vida. El diagnóstico precoz es de gran importancia para poder iniciar el tratamiento a tiempo y evitar una progresión pronósticamente relevante. Las exacerbaciones agudas de la enfermedad pulmonar intersticial pueden complicar aún más el curso. Éstas están asociadas a la progresión de la enfermedad o desencadenadas secundariamente por infecciones.
La enfermedad pulmonar intersticial (EPI) es más común en las colagenosis, especialmente la esclerosis sistémica (SSc), las miopatías autoinmunes, el síndrome de Sjögren, el lupus eritematoso sistémico (LES), pero también en la artritis reumatoide (AR). Durante la evaluación clínica reumatológica, siempre se debe preguntar a la persona afectada sobre la posible presencia de una limitación respiratoria. La presencia de EPI en pacientes con artritis reumatoide limita el pronóstico, la supervivencia y la calidad de vida [1]. El diagnóstico precoz es de gran importancia para poder iniciar el tratamiento a tiempo y evitar una progresión pronósticamente relevante. La progresión de la enfermedad pulmonar intersticial es frecuente y se observa en un grado relevante, sobre todo en pacientes con SSc y AR; recientemente se han publicado criterios para determinar la progresión [2]. Las exacerbaciones agudas de la enfermedad pulmonar intersticial pueden complicar aún más el curso. Éstas están asociadas a la progresión de la enfermedad o desencadenadas secundariamente por infecciones.
Junta del ILD
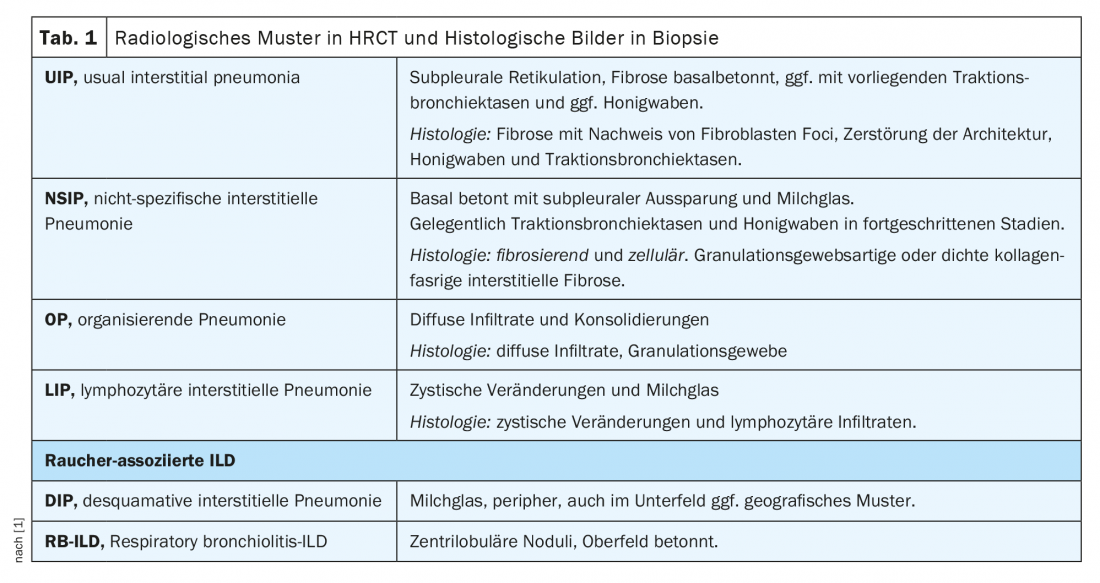
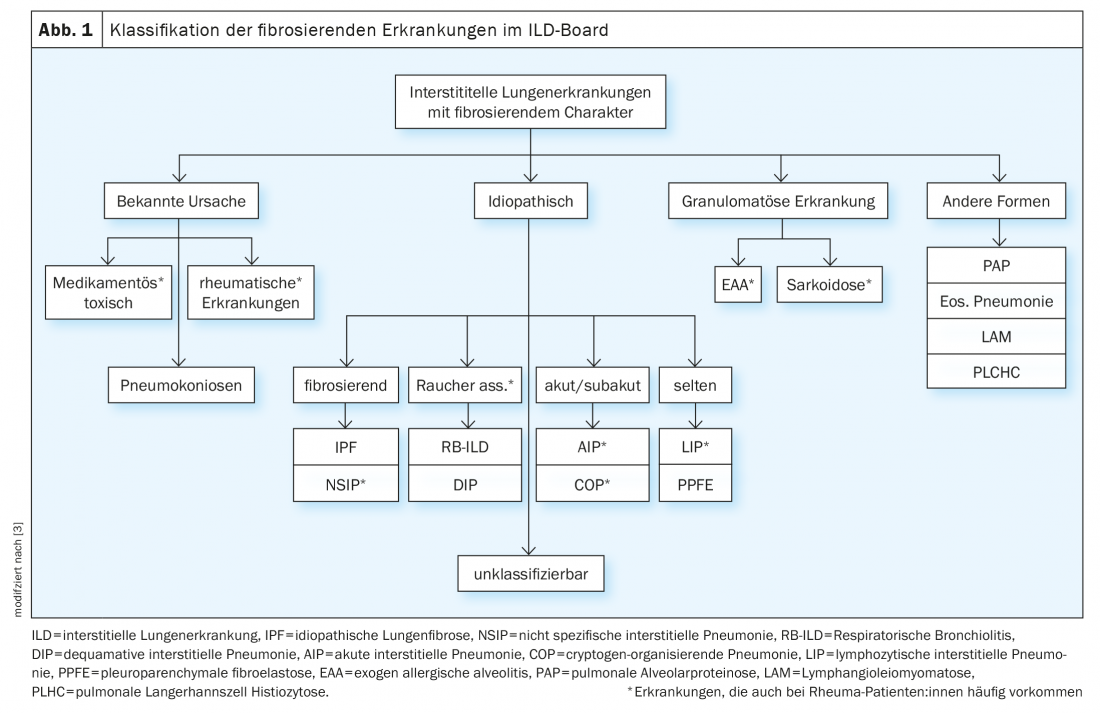
En el marco de la junta de enfermedades pulmonares intersticiales, los casos se discuten de forma interdisciplinar en función de los siguientes parámetros: síntomas principales, antecedentes previos incl. Exposiciones y noxas, serología inmunológica, limitación funcional pulmonar en pletismografía corporal (esp. FVC, TLC, FEV1) y capacidad de difusión (DLCO), diagnóstico por imagen mediante tomografía computarizada de alta resolución del tórax (HRCT), así como resultados de diagnósticos invasivos mediante broncoscopia, incl. Microbiología, hallazgos del lavado broncoalveolar (BAL) y hallazgos histológicos de las biopsias (Tab. 1). Sin embargo, los pacientes reumáticos pueden desarrollar una enfermedad pulmonar intersticial no sólo como consecuencia de la enfermedad reumática, sino también debido a noxas, medicación o exposición (especialmente en el caso del BAL linfocítico), etc. (Fig. 1). Cabe señalar que la obtención de biopsias rara vez está estrictamente indicada en las enfermedades reumáticas confirmadas, pero sirve para simplificar la diferenciación de los diagnósticos diferenciales, sobre todo en las vasculitis y, sobre todo, en la coexistencia de enfermedades tumorales y de otro tipo.
Enfermedades reumáticas inflamatorias con posible afectación de la EPI
RA-ILD: La AR es la enfermedad reumática más común. La presencia de artritis, especialmente en las articulaciones pequeñas, así como de factores reumatoides elevados y anticuerpos contra los péptidos cíclicos citrulinados (la denominada seropositividad) simplifican el diagnóstico y son, por tanto, indicativos [4]. La manifestación extraarticular más común de la AR es la EPI, que se observa en el 25-60% de los pacientes con AR en la TCAR y es responsable del 10-20% de las muertes por AR. La RA-ILD adquiere relevancia clínica en aproximadamente el 10% de los afectados. En el 10% de los casos, la afectación pulmonar precede incluso al desarrollo de la artritis [5]. Los factores de riesgo para desarrollar EPI incluyen el sexo masculino, el tabaquismo y la seropositividad. Las imágenes muestran con mayor frecuencia un patrón UIP. En la literatura se describen mutaciones como la del promotor del gen MUC5B. Fumar puede provocar además un agravamiento de la situación pulmonar. La presencia de una combinación de fibrosis pulmonar y enfisema (CPFE ) se asocia a un pronóstico dramáticamente peor. Diferencialmente, debe considerarse la EPI tóxica por fármacos bajo inmunosupresión. Sin embargo, la EPI asociada al metotrexato (MTX) es extremadamente rara (se calcula que un 0,1%); incluso se ha descrito un efecto protector del MTX.
Por lo tanto, el MTX no debe suspenderse en la AR-ILD. La AR-ILD se trata principalmente optimizando la inmunosupresión. Faltan ensayos controlados aleatorios para el tratamiento de la AR-ILD. Las mejores pruebas apoyan el tratamiento de los pacientes con AR y EPI concomitante con abatacept y rituximab [6]. Los resultados del ensayo APRIL (abatacept en AR-ILD) aún están pendientes. Además, recientemente se ha informado de la eficacia de los inhibidores de JAK o los bloqueantes de IL-6 en la AR-ILD en series de casos [7]. En los cursos progresivos (resumen 1), se recomienda iniciar una terapia antifibrótica. El nintedanib está aprobado para este fin en la UE basándose en los resultados del ensayo INBUILD [8]. El pronóstico de los pacientes con AR-ILD es significativamente limitado en general, con una mediana de supervivencia de unos tres años [1].

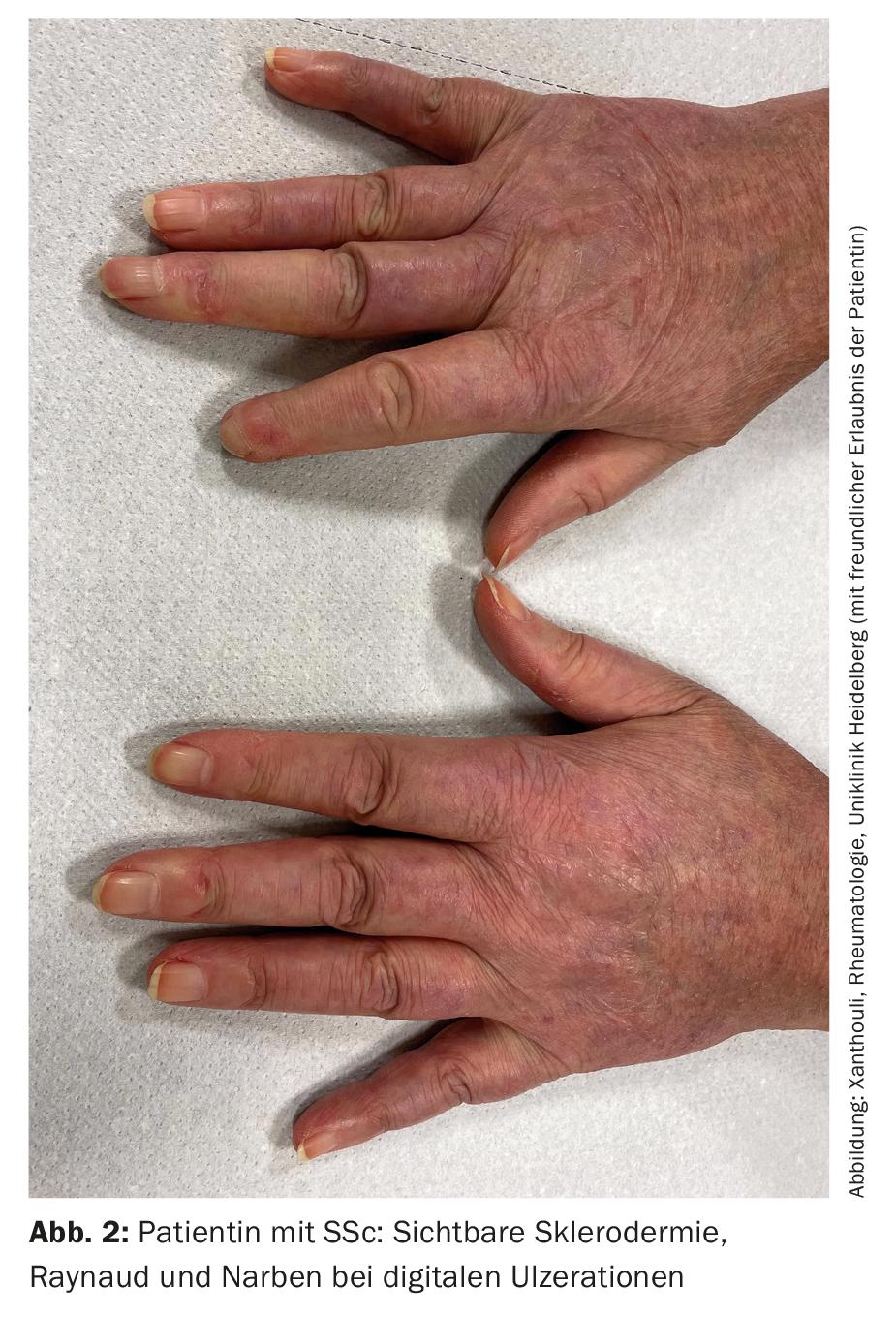
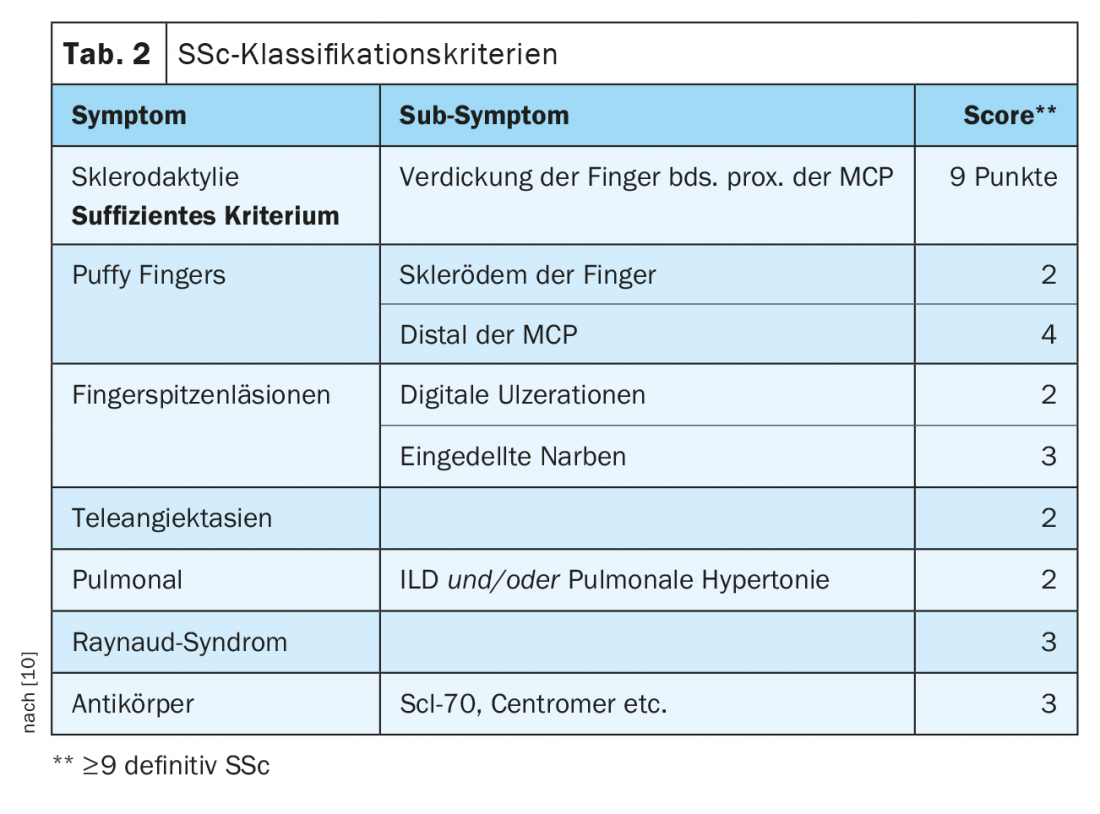
SSc-ILD: Alrededor del 50-85% de los pacientes con SSc ya presentan cambios pulmonares intersticiales en el momento del diagnóstico inicial. En el 20-30% de los casos, esto es progresivo en el curso de la enfermedad. La enfermedad pulmonar intersticial es actualmente la causa más común de muerte en pacientes con SSc, con una mortalidad a los 10 años de aproximadamente el 40% [1]. El diagnóstico correcto y el reconocimiento precoz de la enfermedad pulmonar intersticial son relevantes desde el punto de vista pronóstico [9]. Se han establecido criterios de clasificación para simplificar el diagnóstico (Tab. 2) [10]. La esclerodactilia, la ulceración digital, los síntomas de Raynaud y la microstomía son signos clínicos que deben sugerir inmediatamente la presencia de SSc (Fig. 2). El desarrollo y sobre todo la progresión de la enfermedad se producen predominantemente durante los tres primeros años de la enfermedad. A menudo, la enfermedad pulmonar intersticial es asintomática en la SSc. Se recomienda iniciar el cribado mediante TCAR en el momento del diagnóstico inicial de SSc. Las pruebas de función pulmonar (pletismografía corporal y mediciones de la capacidad de difusión) son necesarias cada trimestre o medio año. Los factores de riesgo para el desarrollo de la enfermedad pulmonar intersticial son el sexo masculino, la presencia de esclerosis sistémica cutánea difusa, la detección de autoanticuerpos contra la Scl-70 y el origen afroamericano [11]. El reflujo, la afectación cutánea difusa (y, por tanto, una puntuación de piel de Rodnan modificada más alta) y el sexo masculino son factores de riesgo de progresión rápida de la enfermedad pulmonar intersticial. En la TC, lo más frecuente es un patrón NSIP, mientras que el patrón UIP se encuentra con menos frecuencia.
El tratamiento es principalmente inmunomodulador con antifibrótico complementario [1]. Basándose en los resultados de los estudios SLS-I y -II, la terapia con ciclofosfamida o micofenolato mofetilo puede iniciarse en SSc-ILD [12,13]. En el estudio FocuSSed, la CVF mejoró en el fenotipo SSc inflamatorio con tocilizumab (anticuerpo contra el receptor de IL-6). Por ello, el tocilizumab ha recibido la aprobación de la FDA para la indicación SSc-ILD, pero se sigue utilizando fuera de etiqueta en Europa [14]. En cuanto al rituximab (también aún fuera de etiqueta), actualmente se dispone de buenos datos para el tratamiento de la SSc-ILD procedentes del estudio DESIRES [15]; los resultados finales del estudio RECITAL aún están pendientes. El estudio SCENCIS es el primer ensayo controlado, aleatorizado, doble ciego y de mayor tamaño que evalúa la eficacia del tratamiento antifibrótico con nintedanib en pacientes con SSc-ILD [16]. El nintedanib está aprobado por la FDA para el tratamiento de la SSc-ILD, pero sólo funciona para prevenir la progresión de la afectación pulmonar y no es adecuado para el tratamiento de otras manifestaciones de la SSc.
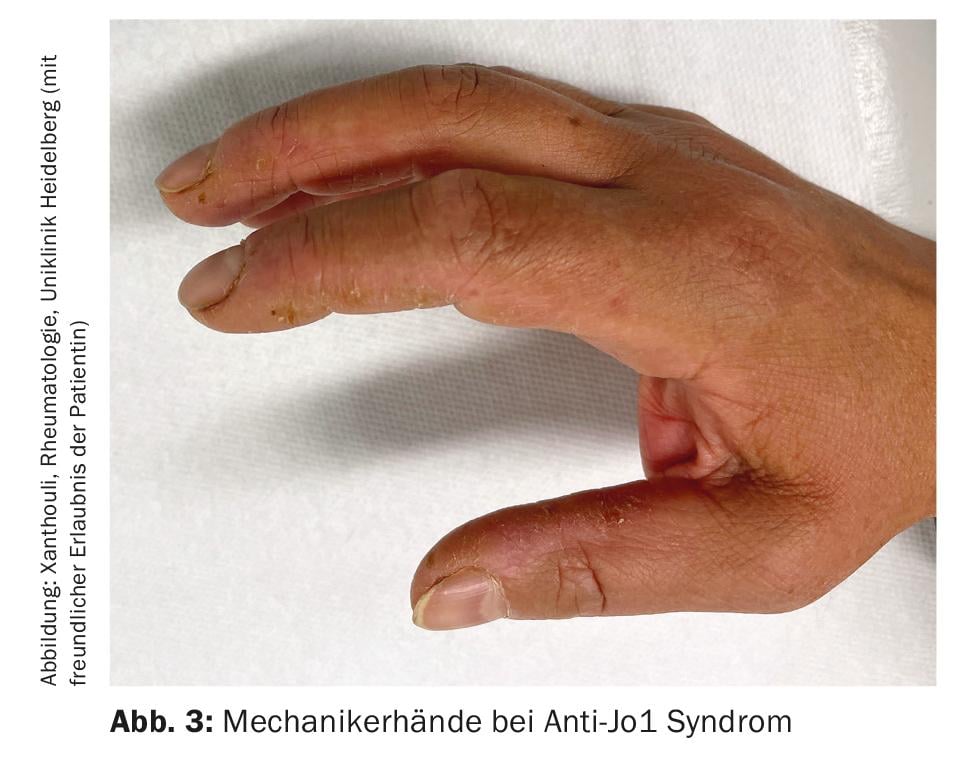
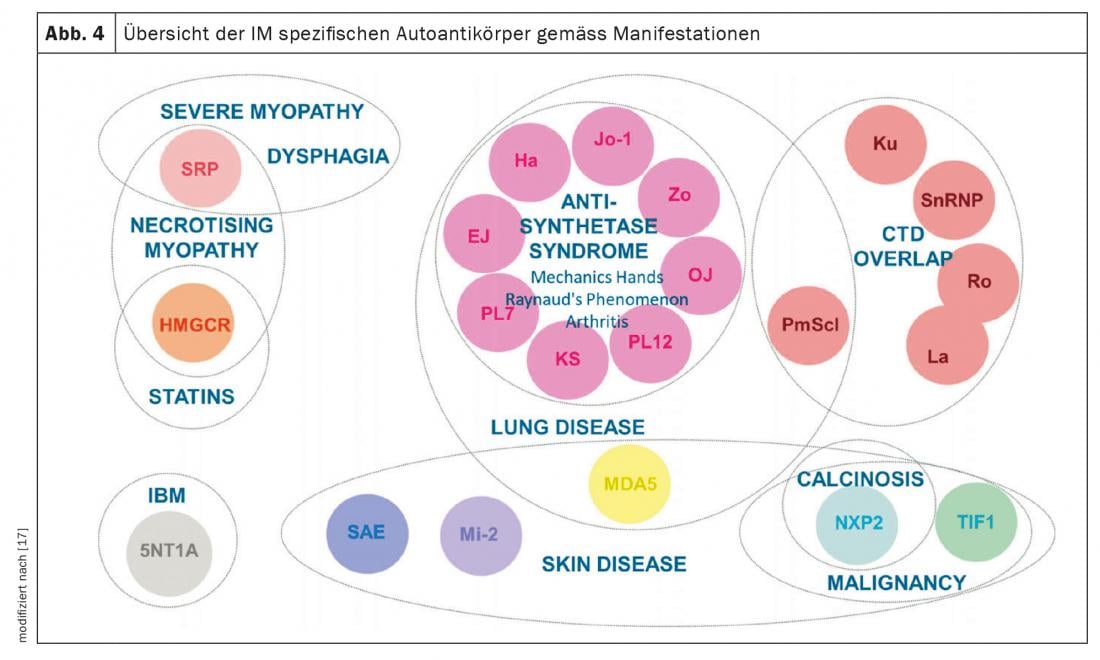
Miositis inflamatoria (MI): La MI incluye el síndrome antisintetasa (ASD), la dermato- y polimiositis, la miositis por cuerpos de inclusión y los síndromes de solapamiento con otras colagenosis. Se manifiestan con y sin afectación muscular (miositis). Los pacientes con afectación muscular presentan valores musculares excepcionalmente elevados en sangre (CK, creatina quinasa) y muestran señales patológicas en la resonancia magnética muscular y en el examen neurofisiológico. Otras manifestaciones clínicas sugestivas de MI son las manos de mecánico (Fig. 3), especialmente en el síndrome antisintetasa, las artritis, el exantema heliotropo en la dermatomiositis y el fenómeno de Raynaud. Hasta el 70% de los pacientes con ASA desarrollan EPI. Además, la EPI es la primera manifestación de la enfermedad en un tercio de las personas con MI [7]. Los autoanticuerpos más comunes en la ASA son los anticuerpos contra Jo-1, pero se conocen otros anticuerpos, por ejemplo contra PL12, PL7, Mi2, etc., que deben determinarse si los síntomas son típicos. La detección de anticuerposMDA5 (proteína 5 asociada a la diferenciación del melanoma) en caso de afectación pulmonar poco clara indica un curso rápidamente progresivo, refractario a la terapia y asociado a una mayor mortalidad, a menudo también sin miositis. Observe la asociación entre neoplasias y MI. Esto es particularmente alto para la detección de anticuerpos TIFγ y NXP2. En el 30% de los casos existe una neoplasia maligna, por lo que es indispensable un cribado tumoral exhaustivo (Fig. 4) [17].
El tratamiento es principalmente con corticoides a dosis altas con buena respuesta especialmente en pacientes jóvenes, aquellos con cirugía, niveles elevados de CK e infiltrados en vaso de leche en la HRCT. En casos graves, debe considerarse el inicio de una terapia de pulso con ciclofosfamida. El tratamiento con micofenolato, rituximab, tacrolimus, abatacept e inhibidores de JAK sigue estando fuera de etiqueta, aunque están respaldados por buenos resultados de estudios retrospectivos [18]. También cabe señalar que existe una autorización de comercialización para la administración de inmunoglobulinas intravenosas en la miositis en la UE [19]. Los pacientes con anticuerpos Jo-1 y evidencia de anticuerpos anti-Ro-52 han mostrado recientemente una buena respuesta al rituximab en lo que respecta a la afectación de la enfermedad pulmonar intersticial, por lo que debería considerarse como una opción de tratamiento [20].
LES: Las polifacéticas manifestaciones pulmonares del LES conducen a menudo a un diagnóstico tardío. Hasta un 12% de los afectados podrían desarrollar una enfermedad pulmonar intersticial en el curso de la enfermedad. Una manifestación especialmente peligrosa del LES es el síndrome hemorrágico pulmonar con hemoptisis grave y rápido deterioro respiratorio. Por otro lado, una disnea grave, un trastorno restrictivo de la ventilación con apraxia diafragmática sin presencia de EPI hacen sospechar la presencia de un síndrome de contracción pulmonar difícil de tratar. Una ecografía torácica, la medición de la fuerza muscular respiratoria y la espiroergometría pueden ser útiles. El tratamiento de la enfermedad pulmonar intersticial en el LES consiste principalmente en intensificar la inmunosupresión. Los fármacos antifibróticos podrían utilizarse en caso de progresión – en casos raros; una autorización para el nintedanib en la UE [21].
Síndrome de Sjögren: Aunque la mayoría de los pacientes de Sjögren apenas manifiestan síntomas pulmonares, en algunos casos se detecta un trastorno restrictivo de la ventilación en el diagnóstico de la función pulmonar. La presencia de síntomas objetivables de sicca con anticuerpos SSA positivos es decisiva para el diagnóstico de la enfermedad subyacente [22]. En el 15% de los casos, la HRCT muestra un patrón LIP con cambios quísticos difusos y bipulmonares con vaso de leche. La biopsia muestra un infiltrado linfocítico. En caso de masas intratorácicas poco claras en el síndrome de Sjögren primario, debe considerarse el linfoma como diagnóstico diferencial. El diagnóstico precoz de la enfermedad pulmonar intersticial asociada al síndrome de Sjögren es relevante desde el punto de vista pronóstico, con una supervivencia a 5 años del 88,5% [23].
Neumonía intersticial con rasgos autoinmunes (IPAF): Los pacientes de ILD con anticuerpos antinucleares (ANA) anormales suelen acudir al reumatólogo. En un 10-20% de los casos, se trata de hallazgos incidentales en el curso del diagnóstico de cribado de la EPI. En la TCAR, el patrón NSIP es el más común y los afectados se benefician significativamente de los esteroides. Sin embargo, rara vez se presentan otros síntomas indicativos de una enfermedad inflamatoria y reumática. En este caso, es necesaria una atención combinada con un neumólogo y un reumatólogo para identificar a tiempo una manifestación tardía de una enfermedad reumática inflamatoria [7].
Otras enfermedades
Vasculitis: En pacientes con granulomatosis con poliangeítis (GPA), a menudo pueden detectarse granumolomas intrapulmonares. En raras ocasiones puede desarrollarse una enfermedad pulmonar intersticial. Por el contrario, cuando se detectan anticuerpos contra la mieloperoxidasa (MPO-ANCA), suele describirse una EPI con un patrón predominantemente UIP. En este caso, debe realizarse un diagnóstico complementario con respecto a otras manifestaciones orgánicas.
Colagenosis mixta: Se estima que la incidencia de la enfermedad pulmonar intersticial es muy variable, del 47 al 78%. Los individuos afectados tienen ANA y anticuerpos RNP U1 positivos. En la TCAR, puede estar presente un patrón NSIP, un patrón UIP o infiltrados en el sentido de una OP. El tratamiento se realiza principalmente con sustancias inmunomoduladoras [7].
Otros tratamientos no medicinales y profilácticos
Todos los pacientes con enfermedades reumáticas deben recibir las vacunas recomendadas, especialmente contra la COVID-19, la gripe y los neumococos, y actualizarlas periódicamente. En fases avanzadas de la enfermedad pulmonar intersticial, puede producirse insuficiencia respiratoria. La sustitución de oxígeno suele ser necesaria durante el curso, empezando con el ejercicio y posiblemente más tarde en reposo. Para ello, se suele realizar una titulación para determinar la dosis. Las medidas de rehabilitación, como los deportes pulmonares y reumáticos, así como las medidas de rehabilitación hospitalaria o ambulatoria, mejoran la situación clínica y la captación de oxígeno de los afectados por enfermedades reumáticas inflamatorias y siguen siendo recomendables. Si se agotan las opciones terapéuticas, debe considerarse la inclusión en la lista para un trasplante de pulmón. No existen directrices claras sobre el momento exacto de la presentación para la cotización. En la SSc, el trasplante autólogo de células madre es cada vez más importante, especialmente en cursos clínicos graves [24]. Si el trasplante no es una opción, debe considerarse el inicio de medidas paliativas y de alivio de los síntomas [25].
Resumen
Las enfermedades reumáticas pueden manifestarse en el parénquima pulmonar y provocar, entre otras cosas, el desarrollo de una enfermedad pulmonar intersticial (EPI). Las manifestaciones pulmonares pueden preceder a la enfermedad reumática subyacente en aproximadamente el 10% de los casos. Por ello, los reumatólogos participan en la junta interdisciplinar de enfermedades pulmonares intersticiales para contribuir al diagnóstico diferencial y a la determinación del algoritmo de tratamiento. El pronóstico de la OIDP reumatoide es limitado, pero la cooperación entre reumatólogos y neumólogos puede mejorar significativamente la calidad de la atención. Aunque la patogénesis del desarrollo de la EPI sigue sin estar clara, el desarrollo de terapias farmacológicas para la EPI reumatoide está progresando. Tanto el reumatólogo como el neumólogo deben tener sólidos conocimientos y confianza en el uso de sustancias inmunomoduladoras y antifibróticas.
Mensajes para llevarse a casa
- Las enfermedades reumáticas inflamatorias pueden manifestarse en el parénquima pulmonar.
- Es necesaria una estrecha colaboración entre neumólogos y reumatólogos para el diagnóstico y el tratamiento.
- El tratamiento es principalmente inmunosupresor.
- Desde 2020, la sustancia antifibrótica nintedanib ha sido aprobada para el tratamiento de la SSc-ILD basándose en los resultados del ensayo SENSCIS. El fármaco también se utiliza para el tratamiento de la enfermedad pulmonar intersticial progresiva (estudio INBUILD).
Literatura:
- Wijsenbeek M, Cottin V: Espectro de las enfermedades pulmonares fibróticas. N Engl J Med 2020; 383: 958-968; doi: 10.1056/NEJMra2005230.
- Raghu G, Remy-Jardin M, Richeldi L et al: Fibrosis pulmonar idiopática (una actualización) y fibrosis pulmonar progresiva en adultos: Guía oficial de práctica clínica de la ATS/ERS/JRS/ALAT. Am J Respir Crit Care Med 2022; 205: e18-e47; doi: 10.1164/rccm.202202-0399ST.
- Kreuter M, Ladner UM, Costabel U, et al: Diagnóstico y tratamiento de la fibrosis pulmonar. Dtsch Arztebl Int 2021; 118; doi: 10.3238/arztebl.m2021.0018.
- Aletaha D, Neogi T, Silman AJ, et al: 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 2010; 69: 1580-1588; doi: 10.1136/ard.2010.138461.
- Duarte AC, Porter JC, Leandro MJ: El pulmón en una cohorte de pacientes con artritis reumatoide: una visión general de los distintos tipos de afectación y tratamiento. Reumatología (Oxford) 2019; 58: 2031-2038; doi: 10.1093/rheumatology/kez177.
- Mena-Vázquez N, Rojas-Gimenez M, Romero-Barco CM, et al: Predictores de progresión y mortalidad en pacientes con artritis reumatoide prevalente y enfermedad pulmonar intersticial: un estudio prospectivo de cohortes. J Clin Med 2021; 10(4): 874.
- Bastian HKA: Pulmón – Enfermedades pulmonares intersticiales. Act Rheumatol 2021; 46: 544-551.
- Flaherty KR, Wells AU, Cottin V, et al: Nintedanib en las enfermedades pulmonares intersticiales fibrosantes progresivas. N Engl J Med 2019; 381: 1718-1727; doi: 10.1056/NEJMoa1908681.
- Xanthouli P, Hermann W, Hunzelmann N, et al: Enfermedad pulmonar intersticial asociada a la esclerodermia. El Neumólogo 2018; 15: 383-395.
- van den Hoogen F, Khanna D, Fransen J, et al: 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis 2013; 72: 1747-1755; doi: 10.1136/annrheumdis-2013-204424.
- Distler O, Volkmann ER, Hoffmann-Vold AM, et al: Perspectivas actuales y futuras sobre el tratamiento de la enfermedad pulmonar intersticial asociada a la esclerosis sistémica. Expert Rev Clin Immunol 2019; 15: 1009-1017; doi: 10.1080/1744666X.2020.1668269.
- Tashkin DP, Elashoff R, Clements PJ, et al: Ciclofosfamida frente a placebo en la enfermedad pulmonar por esclerodermia. N Engl J Med 2006; 354: 2655-2666; doi: 10.1056/NEJMoa055120.
- Tashkin DP, Roth MD, Clements PJ, et al: Micofenolato mofetilo frente a ciclofosfamida oral en la enfermedad pulmonar intersticial relacionada con la esclerodermia (SLS II): un ensayo aleatorizado controlado, doble ciego y de grupos paralelos. Lancet Respir Med 2016; 4: 708-719; doi: 10.1016/S2213-2600(16)30152-7.
- Khanna D, Lin CJF, Furst DE, et al: Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med 2020; 8(10): 963-974.
- Ebata S, Yoshizaki A, Oba K, et al: Seguridad y eficacia del rituximab en la esclerosis sistémica (DESIRES): un ensayo doble ciego, iniciado por el investigador, aleatorizado y controlado con placebo. The Lancet Rheumatology 2021; 3: E489-E497; doi: 10.1016/S2665-9913(21)00107-7.
- Distler O, Highland KB, Gahlemann M, et al: Nintedanib para la enfermedad pulmonar intersticial asociada a la esclerosis sistémica. N Engl J Med 2019; 380(26): 2518-2528.
- Betteridge Z, McHugh N: Autoanticuerpos específicos de la miositis: una herramienta importante para apoyar el diagnóstico de la miositis. J Intern Med 2016; 280: 8-23; doi: 10.1111/joim.12451.
- Mehta P, Aggarwal R, Porter JC, et al: Gestión de la enfermedad pulmonar intersticial (EPI) en los síndromes de miositis: Una guía práctica para clínicos. Best Pract Res Clin Rheumatol 2022; 101769; doi: 10.1016/j.berh.2022.101769.
- Aggarwal R, Charles-Schoeman C, Schessl J, et al: Estudio de fase III prospectivo, doble ciego, aleatorizado y controlado con placebo que evalúa la eficacia y la seguridad de octagam 10% en pacientes con dermatomiositis (“Estudio ProDERM”). Medicine (Baltimore) 2021; 100: e23677; doi: 10.1097/MD.0000000000023677.
- Bauhammer J, Blank N, Max R, et al: Rituximab en el tratamiento del síndrome antisintetasa asociado a anticuerpos Jo1: Positividad anti-Ro52 como marcador de gravedad y respuesta al tratamiento. J Rheumatol 2016; 43: 1566-1574; doi: 10.3899/jrheum.150844.
- Medlin JL, Hansen KE, McCoy SS, et al: Manifestaciones pulmonares en el lupus eritematoso sistémico tardío frente al precoz: Una revisión sistemática y metaanálisis. Semin Arthritis Rheum 2018; 48: 198-204; doi: 10.1016/j.semarthrit.2018.01.010.
- Shiboski CH, Shiboski SC, Seror R, et al: 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjogren’s Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol 2017; 69: 35-45; doi: 10.1002/art.39859.
- Gao H, Zhang XW, He J, et al: Prevalencia, factores de riesgo y pronóstico de la enfermedad pulmonar intersticial en una gran cohorte de pacientes chinos con síndrome de Sjogren primario: un estudio de casos y controles. Medicina (Baltimore) 2018; 97: e11003; doi: 10.1097/MD.0000000000011003.
- Farge D, Ait Abdallah N, Marjanovic Z, et al: Trasplante autólogo de células madre en la esclerodermia. Press Med 2021; 50: 104065; doi: 10.1016/j.lpm.2021.104065.
- Kreuter M, Bendstrup E, Russell AM, et al: Cuidados paliativos en la enfermedad pulmonar intersticial: vivir bien. Lancet Respir Med 2017; 5: 968-980; doi: 10.1016/S2213-2600(17)30383-1.
PRÁCTICA GP 2022; 17(9): 4-9









