En una entrevista concedida a InFo ONKOLOGIE & HÄMATOLOGIE, el Prof. Dr. Christoph Driessen, médico jefe del Departamento de Oncología/Hematología del Hospital Cantonal de St. Gallen, ofrece información sobre la patogenia, el diagnóstico y la terapia en el campo de los linfomas no hodgkinianos (LNH). En particular, analiza el mieloma múltiple y habla de los objetivos del tratamiento y las innovaciones de la investigación que definirán la futura terapia farmacológica.
Prof. Driessen, ¿qué papel desempeñan los virus oncogénicos y las bacterias en el desarrollo del linfoma no hodgkiniano (LNH)?
Prof. Driessen: Disponemos de relativamente poca información confirmada al respecto. Sabemos que algunos LNH están asociados a enfermedades víricas y también encontramos genomas víricos, por ejemplo el virus de Epstein-Barr (VEB), en diferentes células en enfermedades Hodgkin y no Hodgkin con diferentes frecuencias. Pero no podemos decir que el desarrollo de la enfermedad tenga su origen en infecciones víricas.
En cuanto a las bacterias, no existen pruebas definitivas de una asociación de ontogenia entre los linfomas y las enfermedades bacterianas en nuestra zona geográfica.
¿Qué métodos de examen histológico y de imagen son obligatorios para obtener un diagnóstico confirmado?
Hay que distinguir dos cosas: Tenemos que hacer un diagnóstico per se y un diagnóstico de propagación.
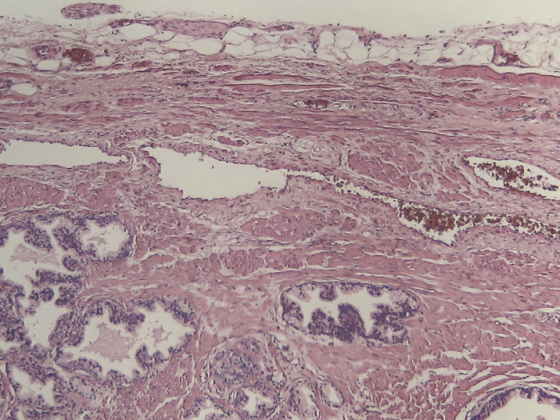
Para el diagnóstico, todavía se necesita una histología concluyente, es decir, normalmente hay que obtener un ganglio linfático o tejido. Una aspiración con aguja fina no suele ser suficiente.
Para el diagnóstico de la diseminación, un procedimiento de imagen seccional de cuerpo entero sigue siendo sin duda el estándar para el LNH. Suele tratarse de una tomografía computarizada, a veces de una resonancia magnética. El examen PET es muy sensible para el diagnóstico de la propagación y está adquiriendo cada vez más importancia clínica. Por el momento, sin embargo, no se trata de un procedimiento de diagnóstico estándar absolutamente necesario. En casos poco claros, es sin embargo un procedimiento muy útil. Además, un examen de la médula ósea mediante citología de médula ósea e histología es estándar para el LNH. Sin embargo, al menos en el LNH altamente maligno, se ha demostrado claramente que si se realizó un diagnóstico por PET y éste fue negativo con respecto a la señal de la médula ósea, se puede prescindir de la citología/histología de médula ósea.
En realidad, el diagnóstico por imagen del SNC y el examen del LCR no se realizan como práctica habitual si, en primer lugar, el paciente no tiene una clínica adecuada que lo sugiera o, en segundo lugar, no pertenece a ninguna población de riesgo particular en la que se sepa que los incidentes del SNC son frecuentes.
¿En qué formas y con la ayuda de qué métodos de tratamiento existe la posibilidad de una curación completa?
En principio, existe una posibilidad de curación para el LNH altamente maligno y de crecimiento rápido. Por lo tanto, la terapia se lleva a cabo con el objetivo de una curación definitiva, lo que incluye la poliquimioterapia junto con un anticuerpo. En la mayoría de las enfermedades de LNH, se utiliza aquí el régimen R-CHOP o terapias relacionadas. Esto significa que se puede lograr una curación definitiva en la mayoría de los casos. Si no funciona, es posible la curación mediante quimioterapia a dosis más altas o de otro tipo junto con un trasplante autólogo de células madre, incluso en la situación de recaída. El trasplante alogénico también es un procedimiento que puede lograr curas definitivas, pero sólo en casos muy seleccionados, normalmente después de que hayan fracasado varias terapias.
En el caso del LNH de baja malignidad, la terapia primaria no tiene pretensión curativa, es decir, el objetivo principal no es curar al paciente de la enfermedad para siempre. Sin embargo, los tratamientos utilizados son similares. Sólo en casos seleccionados existe la posibilidad de lograr a pesar de todo una curación mediante un trasplante alogénico de células madre, pero con riesgos individuales muy grandes.
Un linfoma no Hodgkin agresivo de los linfocitos B es el mieloma múltiple. Aquí se pueden caracterizar diferentes tipos (con proteínas IgG, IgA, IgD, Bence-Jones). ¿En qué se diferencia la terapia en cada caso?
Los subtipos de mieloma múltiple se definen principalmente en función de su fondo genético, es decir, del cambio en el material genético de estas células. El tipo de inmunoglobulina secretada no tiene importancia para la elección de la terapia.
Basándose en el perfil genético y en los marcadores clínicos y serológicos, el mieloma múltiple puede distinguirse como de riesgo estándar, de riesgo más alto y de riesgo más elevado. Estas distinciones siguen debatiéndose actualmente a nivel internacional. Hay algunos puntos en los que se está seguro de que es de muy alto riesgo, pero para todo lo que está en medio, hay diferentes puntos de vista sobre cómo clasificarlo exactamente. Se están investigando enfoques terapéuticos adaptados al riesgo, pero no tenemos pruebas definitivas de que deba utilizarse una terapia específica y sólo ésta para determinadas constelaciones de riesgo. Una excepción a esto es quizá la translocación (4;14), que sabemos que se ha asociado previamente a un riesgo desfavorable, pero que tiene un resultado de riesgo estándar cuando se utiliza bortezomib (Velcade®) en la terapia de primera línea. Por lo tanto, la constelación de riesgos puede superarse con este fármaco.
El mieloma múltiple no tiene cura. ¿Cuáles son los objetivos realistas que pueden alcanzarse con los métodos terapéuticos actuales?
Por supuesto, los objetivos dependen del paciente y de la enfermedad. En pacientes más jóvenes o en pacientes de hasta 65 años que reúnan los requisitos para recibir una terapia de dosis altas, el objetivo es prolongar la supervivencia estadística con el menor deterioro posible de la calidad de vida como resultado de la terapia. Concretamente, esto significa el intervalo libre de terapia más largo posible tras la terapia primaria y la toxicidad controlada.
Incluso en los pacientes “ancianos aptos” que no reúnen los requisitos para una terapia de dosis altas, el objetivo es prolongar la supervivencia estadística y evitar las complicaciones relacionadas con el mieloma, manteniendo al mismo tiempo una buena calidad de vida. Para ello, sin embargo, no se utiliza el tratamiento de altas dosis, sino nuevos fármacos en combinación. Así, se obtienen resultados al menos numéricamente similares.
La tercera categoría son los pacientes “mayores-no aptos”. Aquí ya no podemos hacer una terapia más intensiva debido a consideraciones de toxicidad. El objetivo aquí es claramente evitar las complicaciones relacionadas con el mieloma, pero también las relacionadas con la terapia, y optimizar la calidad de vida, pero no prolongar la supervivencia global. Por lo tanto, se aplica un tratamiento poco tóxico.
¿Existen innovaciones o enfoques de investigación en el campo de la terapia del mieloma múltiple que inspiren esperanza?
Hay muchos enfoques que dan motivos para la esperanza. Por un lado, tenemos los nuevos fármacos que se han introducido, como la lenalidomida (Revlimid®) y el bortezomib (Velcade®), todos los cuales tienen ahora fármacos sucesores de los mismos grupos de sustancias que son menos tóxicos y, en algunos casos, siguen funcionando cuando los fármacos de primera generación ya no lo hacen. Algunos de estos medicamentos han sido aprobados en EE.UU., otros ya han sido aprobados por la EMA y también estarán disponibles en Suiza en un futuro próximo. En el contexto del tratamiento estándar, todavía no lo son.
Además, hay muchos anticuerpos específicos contra el mieloma en desarrollo que llegarán al mercado en los próximos años. Espero un cambio en el panorama terapéutico similar al que se produjo con la introducción del rituximab.
Las inmunoterapias están haciendo grandes progresos. En la terapia del mieloma nos encontramos con la situación de que conseguimos un control muy bueno de la enfermedad al principio, pero no podemos controlar la enfermedad mínima residual. En realidad, éste es el mejor campo de aplicación de las inmunoterapias y me parecen muy alentadores los éxitos que ya se han logrado aquí y que pueden esperarse en los próximos años.
Todos estos nuevos fármacos, la mayoría de los cuales son orales, tienen relativamente pocos efectos secundarios y son una opción válida para los tres grupos de pacientes descritos anteriormente.
¿En qué pacientes está indicado el trasplante autólogo o alogénico de células madre?
El trasplante autólogo de células madre con altas dosis de quimioterapia sigue siendo la principal terapia de elección para los pacientes que son biológicamente capaces de hacerlo, normalmente los menores de 60 años, a veces menores de 70.
El trasplante alogénico de células madre en el mieloma, al igual que en otros mielomas poco malignos, es una forma de terapia que, a diferencia de otras opciones de tratamiento, ofrece potencialmente una posibilidad de curación, pero se asocia a riesgos individuales muy elevados. En general, se es bastante cauto con la forma alogénica en el mieloma múltiple porque los efectos inmunológicos conseguidos con la médula ósea ajena no son tan buenos como en otros linfomas poco malignos.
Por el momento, parece que algunos pacientes con mieloma múltiple de alto riesgo pueden beneficiarse del trasplante autólogo/alogénico en tándem en la terapia primaria.
Entrevista: Andreas Grossmann
InFo Oncología y Hematología 2013; 1(1): 30-32
PRÁCTICA GP 2014; 9(1): 61-63