La esclerosis sistémica (SSc) es una de las enfermedades más graves porque está presente en todo el cuerpo, pero hay solución para pocos problemas y prácticamente cada órgano debe tratarse individualmente. Aún no existe una medicación universal para ellas como para otras enfermedades, es decir, ni un biológico ni un inmunosupresor específico.
Un programa obligatorio para todo paciente que presente esclerosis sistémica es el examen de los pulmones, por dos componentes: uno es la fibrosis pulmonar, que es peor, y el otro es la hipertensión pulmonar (HAP), para la que existen al menos más fármacos. La pérdida de extensibilidad de los pulmones y la pérdida de su función de intercambio gaseoso son los dos patomecanismos del patrón de enfermedad de la fibrosis pulmonar. Es 100 veces más rara, pero provoca más muertes al año que el asma. El dilema es que existen más de cien afecciones diferentes que pueden conducir a la fase final de la fibrosis pulmonar.
“El deterioro puede ser muy rápido en la esclerosis sistémica, y eso es lo peligroso”, advirtió el profesor Dr. Ulf Müller-Ladner, del Departamento de Reumatología e Inmunología Clínica de la Clínica Kerckhoff de Bad Nauheim (D). “Los pacientes no presentan grandes signos, quizá una úlcera digital, y de repente la fibrosis pulmonar o la hipertensión pulmonar ya están ahí”. Los propios pacientes sólo se dan cuenta cuando comienzan los síntomas de disnea. Por lo tanto, según el reumatólogo, también es elemental en su disciplina examinar siempre los pulmones de un paciente, independientemente de que muestre síntomas o no.
En la SSc, la fibrosis pulmonar ha adquirido cada vez más importancia en la lista de manifestaciones orgánicas. No en vano, otras complicaciones como las crisis renales o los problemas cardiovasculares o gastrointestinales se han vuelto más tratables con el paso del tiempo, no así la fibrosis pulmonar asociada a la SSc (SSc-ILD). “Si se estratifica en función de las dimensiones en la HRCT, los pacientes con SSc muestran fibrosis en la TC en más del 30% de los casos”, afirmó el profesor Dr. Andreas Günther, Jefe del Foco de Enfermedades Pulmonares Fibrosantes del Hospital Universitario Giessen Marburg. “Así que ya es una enfermedad más grave y con peor pronóstico”.
Mejor seguimiento = resultado más controlado
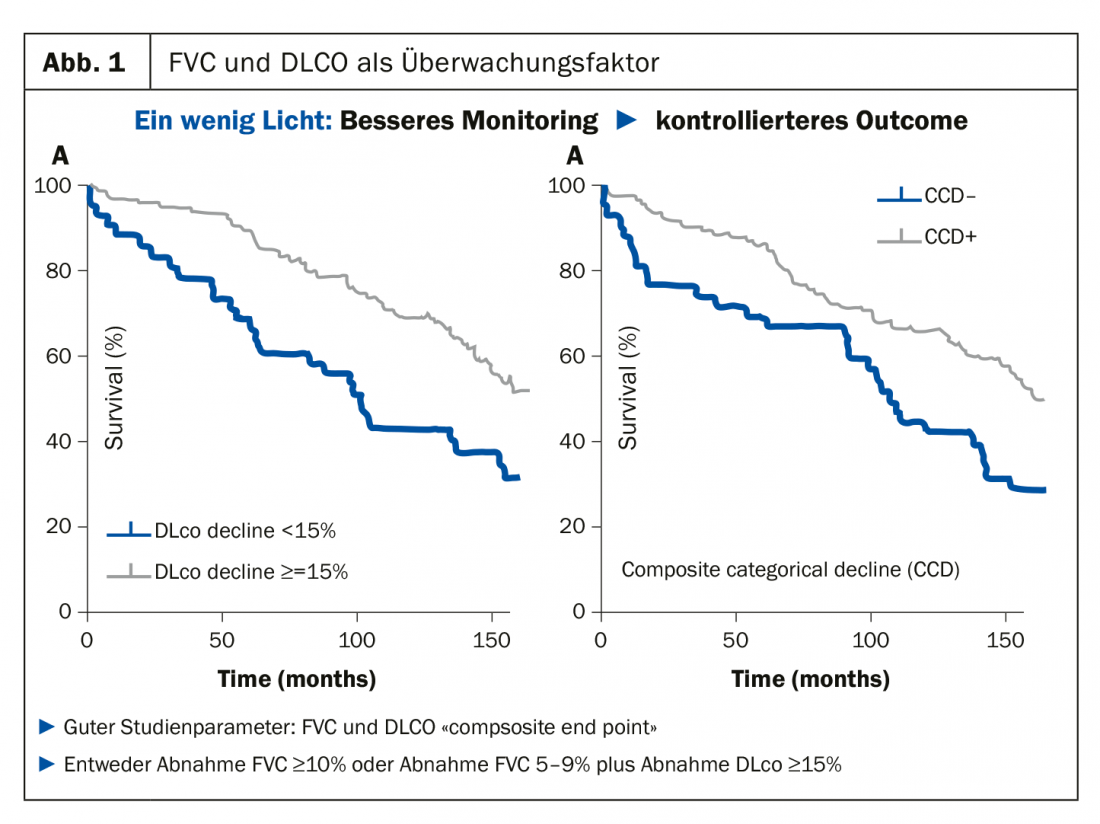
Como reumatólogo que tiene que vigilar y monitorizar los pulmones, el Prof. Müller-Ladner siempre aconseja a sus colegas que se ciñan a los parámetros relativamente fáciles de medir, es decir, la capacidad vital (CVF) y la capacidad de difusión (DLCO), ya que esta combinación en particular ha evolucionado muy positivamente como factor de monitorización en los estudios. “Si la combinación de estos dos parámetros progresa muy rápidamente, entonces las curvas también divergen” (Fig. 1). Este punto final compuesto está ahora también establecido en los estudios. Una disminución de la CVF de ≥10% o una disminución del 5-9% más una disminución de la DLCO de ≥15% deben considerarse decisivas. “Como reumatólogos, podemos sonreír ante estas pequeñas cifras, pero en relación con los pulmones, son absolutamente valiosas”, el especialista dejó claro un factor que sus colegas tienden a subestimar: “Un pequeño cambio en los pulmones puede tener un gran impacto, y los pacientes también lo notan”.
“Lo principal es hacer algo”
La terapia para muchas enfermedades reumatológicas es similar: hay células inflamatorias, que se inmovilizan, y luego todo va bien – los efectos secundarios se dejan a un lado. La situación es diferente con el SSc. “Porque aquí tienen lugar varios componentes en paralelo y nadie sabe muy bien con qué empieza: ¿el sistema inmunitario, la fibrosis, el vascular?”. Y si baja uno de estos interruptores, puede utilizarlo para subir algo en otro lugar, lo que no mejora el estado general. Al mismo tiempo, uno se enfrenta a la fibrosis, “y antagonizar la fibrosis es una de las tareas más difíciles de todas”. Así que la pregunta sigue siendo cómo proceder en el tratamiento: ¿eliminar el desencadenante o dirigir la unión?
Los reumatólogos responden a esta pregunta haciendo lo que pueden y saben, según el Prof. Müller-Ladner: toman metotrexato o penicilamina, “lo principal es hacer algo”. Pero a veces el accionismo también puede causar daños, por ejemplo si el diagnóstico no es correcto. Si los hallazgos no están claros, dice el experto, siempre se debe consultar a un neumólogo para estar seguros. El profesor Günther se refirió a las directrices de tratamiento EULAR/EUSTAR SSc-ILD de 2016, que recomiendan la ciclofosfamida o el trasplante autólogo de células madre hematopoyéticas (HSCT) (Tab. 1). El micofenolato mofetilo (MMF), según el neumólogo, no es ciertamente menos eficaz que la ciclofosfamida, sino menos tóxico, razón por la cual se sigue prescribiendo a menudo.

Estudios prometedores
Otro problema: la esclerosis sistémica es y sigue siendo una enfermedad rara, lo que hace que los grandes estudios sean deseables pero escasos. “Sacudírsela de la muñeca como se hace con la artritis reumatoide (AR) no funciona porque simplemente no le salen las cuentas”, explicó el Prof. Müller-Ladner. Por eso cada publicación es valiosa. El reumatólogo presentó en primer lugar dos estudios (FaSScinate y FocuSSced) con un número relativamente pequeño de participantes, ambos centrados en combatir la inflamación con interleucina-6 (IL-6) utilizando tocilizumab. Especialmente en el estudio FocuSSced, se vio que en el rango a largo plazo, la antiinflamación relacionada con la CVF bajo IL-6 era prometedora.
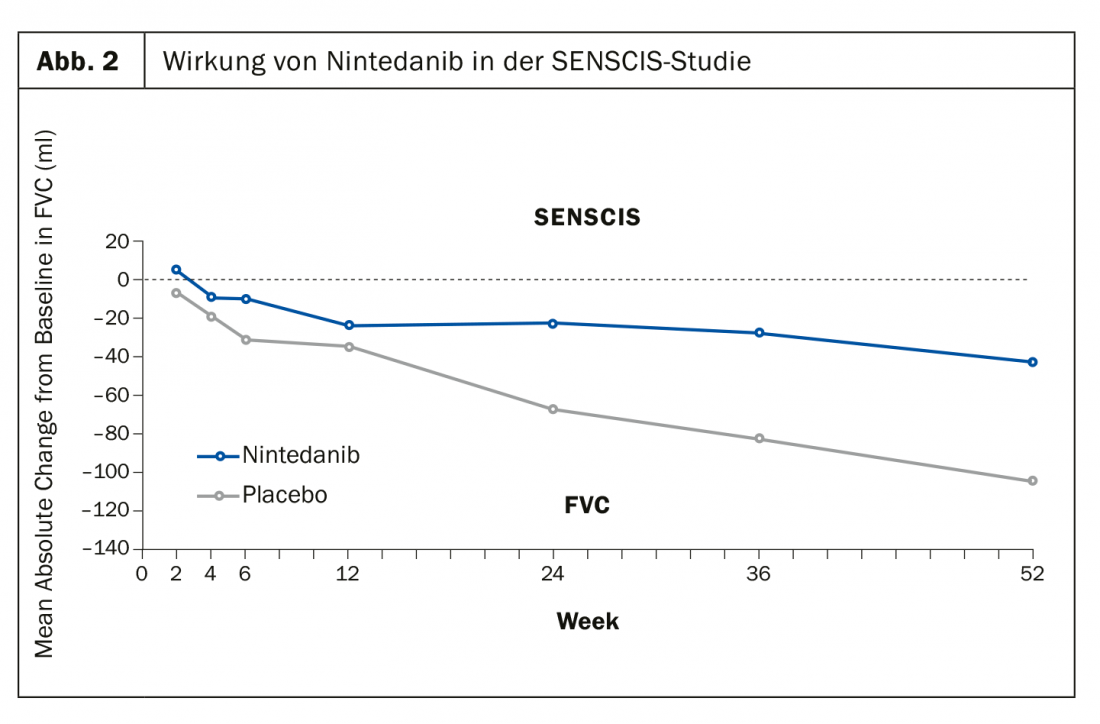
El estudio SENSCIS, por su parte, investigó el efecto del nintedanib frente al placebo con el criterio de valoración primario de la reducción de la capacidad vital funcional (mL/año). Se orienta hacia una medicación básica ajustada de forma estable en la vida real con MTX o MMF. También en este caso los resultados fueron positivos después de 52 semanas (Fig. 2). “Necesitamos un fármaco antifibrótico sensato”, confía el Prof. Müller-Ladner, “porque hasta ahora la fibrosis sólo puede tratarse indirectamente”.
Sin embargo, el nintedanib no está aprobado actualmente para el tratamiento de la SSc/SSc-ILD u otra enfermedad pulmonar intersticial progresiva (PF-ILD) fuera de la fibrosis pulmonar idiopática (FPI). Esto hace que sea aún más importante obtener la aprobación en un futuro próximo no sólo para las formas fibróticas puramente pulmonares de la enfermedad.
Fuente: Simposio industrial “Fibrosis pulmonar: el reto de la SSc y otras enfermedades reumáticas” como parte del DGRh; organizador: Boehringer Ingelheim
InFo NEUMOLOGÍA Y ALERGOLOGÍA 2019; 3(1): 30-31 (publicado el 11 de diciembre de 19, antes de impresión).