La terapia génica, la vieja esperanza de la investigación médica, se abre paso por fin en la clínica para curar las enfermedades monogenéticas. Así, la medicina tiene a su disposición una nueva terapia que puede tratar las enfermedades hereditarias no sólo sintomática sino también causalmente. Otro enfoque de la terapia génica es el tratamiento de las alteraciones génicas adquiridas en neoplasias malignas mediante células T CAR (receptor de antígeno quimérico).
La terapia génica, la vieja esperanza de la investigación médica, se abre paso por fin en la clínica para curar las enfermedades monogenéticas. Así, la medicina tiene a su disposición una nueva terapia que puede tratar las enfermedades hereditarias no sólo sintomática sino también causalmente. Otro enfoque de la terapia génica es el tratamiento de las alteraciones génicas adquiridas en neoplasias malignas mediante células T CAR (receptor de antígeno quimérico). A continuación se ofrece una visión general de los principios biológicos moleculares y de la aplicación clínica de los procedimientos actuales de terapia génica y celular.
Los genes como planos de todas las proteínas – las mutaciones como causa de las enfermedades monogenéticas
Cada célula humana contiene un plano genético dentro del núcleo celular en forma de ácido desoxirribonucleico (ADN). El ADN está formado por cuatro componentes básicos, las bases adenina (A), timina (T), guanina (G) y citosina (C), que, alineados en largas cadenas lineales, forman la estructura básica de los cromosomas de una célula humana. El ADN de cada célula contiene unidades de codificación individuales, los genes. Cada gen consta de una región codificante y una unidad reguladora, el promotor, cuya actividad puede ser amplificada por un elemento potenciador si es necesario. Cada gen codifica la estructura de una proteína a través de la secuencia de los cuatro componentes básicos del ADN. Las proteínas se componen de 20 elementos básicos, los aminoácidos, que se pliegan en una o varias cadenas lineales para formar proteínas. Sólo las proteínas correctamente plegadas pueden desempeñar su función biológica en la célula.
Enfermedades monogenéticas
Si se produce una reparación defectuosa tras un daño en el ADN, puede alterarse la secuencia de los componentes básicos del ADN. Alternativamente, también pueden perderse los componentes básicos del ADN. Estos cambios se denominan “mutaciones”. Si se produce una mutación en una sección del ADN que codifica una proteína, esto puede alterar la estructura de la proteína codificada en ella: La proteína defectuosa sintetizada según el plano de ADN no puede cumplir su función natural en la célula o sólo puede hacerlo de forma incompleta. Este cambio genético, en el caso más simple por ejemplo un intercambio de una A por una C dentro del ADN, puede ser la causa causal de una enfermedad genética, si la función de la célula en un tejido se reduce o incluso falla como resultado, por ejemplo.
- Cuando el metabolismo en las células funciona de forma insuficiente y se acumulan metabolitos (por ejemplo, defecto de la lipoproteína lipasa).
- Cuando la síntesis de los componentes básicos individuales por parte del metabolismo es deficiente (por ejemplo, defecto de la glucosa-6-fosfato deshidrogenasa).
- Si los componentes individuales del sistema inmunitario no funcionan (por ejemplo, granulomatosis séptica o inmunodeficiencia combinada grave).
En un estudio reciente, 4166 enfermedades raras monogenéticas podrían estar relacionadas causalmente con 3163 genes [1]. La cura de la enfermedad de causa genética sólo es posible si es posible compensar o corregir la causa causal.
Nuevos enfoques para la curación a través de la terapia génica
Si se ha identificado una mutación genética claramente definida como causa de una enfermedad, ésta se convierte en accesible para una potencial terapia génica. Existen varios procedimientos disponibles:
- En las enfermedades en las que la mutación del gen conduce a la pérdida de expresión de la proteína o a la expresión de una proteína defectuosa (mutaciones de “pérdida de función” o “disfunción”), puede realizarse la adición del gen.
- En las enfermedades en las que la mutación genética conduce a una sobrefunción de la proteína (mutaciones de “ganancia de función”), se puede intentar la reparación genética.
En la adición de genes, el genoma de la célula se amplía con un gen que codifica correctamente la proteína que falta o que funciona mal y compensa así la función del gen mutado. El resultado es el restablecimiento de la función original de la proteína. La adición de genes se ha probado en ensayos clínicos para el tratamiento de numerosas enfermedades, con éxito clínico en más de 20 enfermedades hereditarias congénitas. Todos los productos de terapia génica aprobados actualmente se basan en la adición de genes.
En la reparación de genes con nucleasas como CRISPR-Cas9 (fusión de la nucleasa Cas9 con CRISPR de unión al ADN, repeticiones palindrómicas cortas agrupadas y regularmente espaciadas), la secuencia correcta de ADN se restaura en la célula dirigiéndose a la mutación y corrigiéndola in situ (principio: cortar y reemplazar). De este modo, se restablece la función prevista de la proteína y, posteriormente, se produce una normalización de la función biológica. El uso de la reparación génica (“tijeras génicas”/CRISPR-Cas) es posible independientemente de si existen mutaciones de “ganancia de función” o de “pérdida de función”. Esta modificación específica de la secuencia de la información genética es posible en el laboratorio, pero hasta ahora sólo se ha utilizado en oncología en el contexto de unos pocos ensayos clínicos.
Genaddition
Los primeros éxitos clínicos mediante terapia génica se lograron en el campo de la inmunología. Las causas de las inmunodeficiencias congénitas residen en las células madre hematopoyéticas. Por ello, los pacientes afectados pueden curarse a menudo mediante el trasplante de células madre hematopoyéticas procedentes de un donante extranjero o familiar (trasplante alogénico). Los familiares rara vez se utilizan como donantes HLA-haploidénticos debido a una mayor tasa de efectos secundarios. Los miembros de la familia que tienen mayor identidad HLA pero también son portadores del defecto genético tienen más probabilidades de no ser elegibles. En ausencia de un donante HLA-idéntico (en aproximadamente 1/3 de los caucásicos, más frecuentemente en otros grupos étnicos), la terapia génica autóloga puede conducir a la curación de la enfermedad.
Para la terapia génica ex vivo, las células autólogas se afinan tras la movilización con G-CSF, se purifican, se enriquecen y se cultivan ex vivo durante un breve periodo de tiempo. Durante este periodo, las células madre se tratan con el vector de terapia génica y después se reinfunden en el paciente, normalmente después de la quimioterapia para ablacionar la médula ósea. Sorprendentemente, dependiendo de la inmunodeficiencia, una corrección parcial puede ser suficiente para una mejora clínica significativa.
La primera publicación sobre el éxito de la terapia génica apareció en el año 2000 sobre el tratamiento de bebés con la forma más grave de inmunodeficiencia congénita, la inmunodeficiencia combinada grave, que suele conducir a la muerte sin trasplante en el primer año de vida debido a las infecciones más graves [2].
Entre 2000 y 2006, todos los éxitos clínicos en el campo de la terapia génica se lograron con la ayuda de los llamados vectores retrovirales. Éstas introducen la secuencia de corrección en forma de ARN en las células madre, donde se convierte en ADN mediante transcripción inversa y después se integra en el ADN del paciente (adición de genes). Posteriormente, se forma la proteína funcional y así se corrige clínicamente el defecto.
Para una adición genética in vivo, las células del cuerpo no se extraen, se tratan y se devuelven, sino que las partículas virales con la secuencia de corrección se inyectan directamente en el cuerpo. La primera adición genética in vivo se publicó en 2007: para el tratamiento de la enfermedad de Parkinson, se inyectó a los pacientes de forma unilateral y subtalámica partículas virales adenoasociadas (AAV) modificadas que codificaban la descarboxilasa del ácido glutámico [3]. La gran mayoría de los ensayos clínicos de terapia génica in vivo utilizan partículas AAV modificadas para introducir información genética en el organismo. Las excepciones son el uso de partículas virales modificadas derivadas del virus del herpes simple-1 (VHS-1) [4] o de partículas adenovirales no replicantes [5] para el tratamiento del glioblastoma y el ensayo de partículas virales derivadas del VIH-1 para el tratamiento in vivo de la enfermedad de Parkinson [6].
Efectos secundarios en los primeros estudios de terapia génica
En la primera generación de terapias génicas ex vivocon vectores γ-retrovirales, se ha demostrado que el lugar de integración del vector de terapia génica en el genoma de la célula diana desempeña un papel clave con respecto a los posibles efectos secundarios. Estos vectores de primera generación se integraron preferentemente cerca de los sitios de inicio de la transcripción. En los vectores de terapia génica γ-retrovirales de primera generación, la expresión de la proteína era impulsada por un promotor en el vector de terapia génica y potenciada por un potenciador. El potenciador del vector de terapia génica de primera generación fue capaz de interactuar con el sitio de inicio de la transcripción del gen en el que se integró el vector de terapia génica. Si se producía la integración en un oncogén, éste podía activarse además de la expresión de la proteína terapéutica, lo que conducía a la curación de la enfermedad subyacente. Como resultado, las células madre en las que se produjo esta constelación habían adquirido una ventaja de crecimiento y eran capaces de multiplicarse clonalmente. En algunos ensayos clínicos con diversas inmunodeficiencias, esto provocó que algunos pacientes desarrollaran neoplasias hematológicas como efecto secundario de la terapia génica [7–10]. Estos efectos secundarios hicieron que a partir de 2009 se pasara de los vectores γ-retrovirales a los vectores lentivirales derivados del virus VIH-1, ya que su perfil de integración promete más seguridad [11]. Además, en los denominados vectores lentivirales autoinactivadores (SIN), se eliminaron el promotor derivado del virus VIH-1 y su elemento potenciador. La ausencia de elementos potenciadores en los vectores de terapia génica de segunda y tercera generación impide la transactivación de oncogenes mediada por potenciadores tras el tratamiento ex vivo decélulas madre. Hasta la fecha, los ensayos clínicos han tratado las células madre de aproximadamente 100 pacientes con vectores lentivirales SIN. Hasta ahora, ninguno de estos ensayos clínicos ha dado lugar al desarrollo de neoplasias hematológicas, lo que indica una mejora significativa de la seguridad en comparación con el vector de primera generación.
Los vectores de terapia génica basados en AAV utilizados para la adición de genes invivo sólo se integran en pequeña medida en el genoma de las células diana. Por lo tanto, la mutagénesis de inserción es poco probable. El mayor potencial de riesgo de los vectores de terapia génica basados en AAV reside en cualquier inmunidad preexistente a las estructuras superficiales del vector AAV. En un ensayo clínico para el tratamiento de la hemofilia B, se utilizó por vía intramuscular un vector AAV2 que codifica el factor IX de la coagulación sanguínea. Una respuesta inmunitaria mediada por células T provocó la eliminación de todas las células modificadas genéticamente y puso fin al efecto terapéutico [12]. Por el contrario, la administración intravenosa produjo la captación de los vectores en los hepatocitos y efectos terapéuticos a largo plazo en pacientes en los que no se pudieron detectar anticuerpos contra el AAV antes de su inclusión en el estudio [13].
Éxitos de la terapia génica
La primera generación de vectores de terapia génica γ-retroviral proporcionó pruebas de que la terapia génica puede conducir al éxito clínico para la X-SCID (inmunodeficiencia combinada grave ligada al cromosoma X) [2], la ADA (adenosina deaminasa)-SCID [14], la X-CGD (granulomatosis séptica ligada al cromosoma X) [9], la epidermólisis bullosa [15] y para el síndrome de Wiskott Aldrich (WAS) [16].
Debido a los efectos secundarios mencionados en el tratamiento de la IDCG-X, la IDCG-X y el WAS con vectores de terapia génica de primera generación (vectores γ-retrovirales con promotor/ potenciador completo), se desarrollaron los vectores lentivirales SIN, que dieron lugar a un éxito clínico. Hasta ahora, se han logrado éxitos clínicos con vectores lentivirales SIN en terapia génica ex vivo para el tratamiento de
- Adrenoleucodistrofia ligada al cromosoma X (ALD) [17]
- γ-Talasemia [18,19]
- QUÉ [20]
- X-SCID [21]
- ADA-SCID [22]
- Anemia falciforme [23]
- COL7A1 Epidermólisis bullosa [24]
- Leucodistrofia metacromática [25]
Mientras tanto, la terapia génica in vivomediada por AAV también se ha utilizado en el tratamiento de al menos 14 indicaciones:
Neurología
- Parkinson [3,26,27]
Oftalmología
- Cong del hígado. Amaurose [28,29]
- Corioideremia [30,31]
- Degeneración macular asociada a la edad [32]
- Neuropatía óptica hereditaria de Leber [33,34]
Hematología
- Hemofilia B [13]
Distrofia muscular
- Distrofia muscular de Becker [35]
- Atrofia muscular espinal tipo I (AME1) [36]
Enfermedades metabólicas
- Enfermedad de Pompe [37]
- Deficiencia de α-1-antitripsina (AAT) [38,39]
- Mucopolisacaridosis tipo IIIB [40]
- Deficiencia de aminoácidos aromáticos descarboxilasa [41,43]
- Deficiencia de lipoproteína lipasa [42]
En el tratamiento de la leucemia, las células CAR-T se utilizan en numerosos estudios y ahora también son terapias aprobadas. Para ello, se introducen receptores de antígenos quiméricos en células T autólogas de pacientes afectados. Esto permite a estas células T modificadas genéticamente (células CAR-T) reconocer y eliminar las células tumorales [44–46].
Reparación de géneros
Hasta ahora, no ha sido posible integrar vectores de terapia génica lenti o γ-retrovirales en lugares predeterminados del genoma. La inserción y/o corrección de secuencias específicas sólo fue posible cuando se pudo cortar el ADN de forma específica para cada secuencia. Para ello se utilizan nucleasas como CRISPR-Cas. En este sistema de terapia génica se ha producido una fusión de las repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas (CRISPR) de unión al ADN y el cortador de ADN Cas9 (CRISPR/Cas9) [47]. La ruptura resultante en el ADN puede entonces ser reparada por la célula de dos maneras diferentes: O bien los extremos se vuelven a unir en un proceso defectuoso denominado unión de extremos no homólogos. El segundo mecanismo de reparación es la recombinación homóloga. Esta vía se toma cuando hay ADN de reparación en la célula cuyos extremos coinciden con la secuencia de ADN en el lugar de la rotura del ADN. Este ADN de reparación se administra a las células junto con la nucleasa y sirve como plantilla genética. En la recombinación homóloga, la rotura del ADN es entonces cerrada por las enzimas endógenas de reparación celular según la secuencia del ADN de reparación. Esto permite introducir en el genoma un gen corrector de una secuencia específica o corregir genéticamente mutaciones puntuales. La adición de genes de secuencia específica aún no ha encontrado aplicación clínica porque la eficacia del método ha sido limitante hasta ahora. Sin embargo, los recientes avances técnicos están haciendo posible la realización de ensayos clínicos.
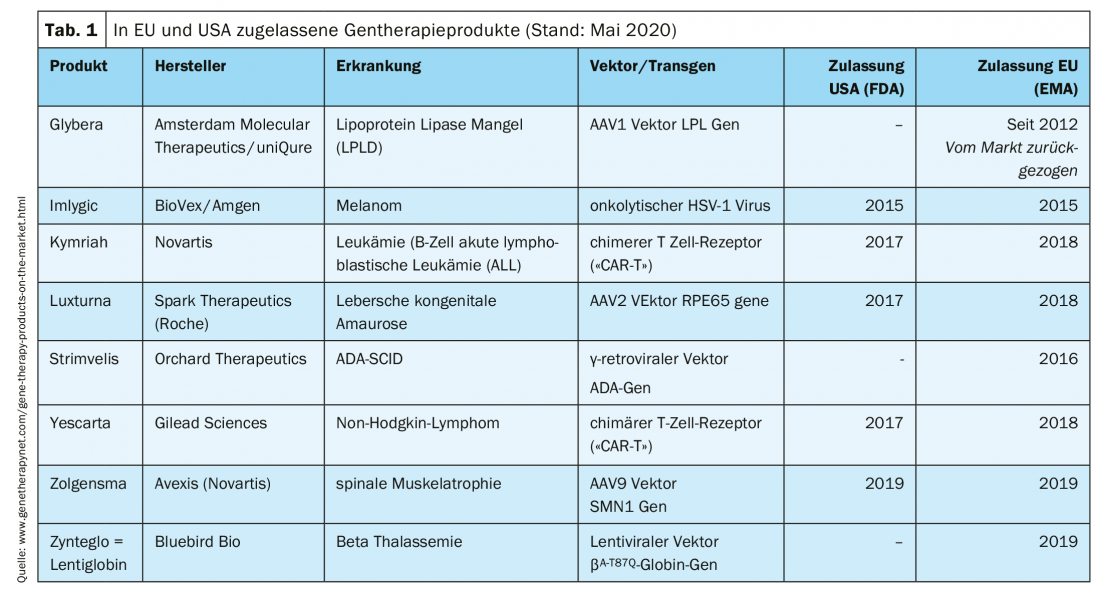
Productos de terapia génica aprobados clínicamente
El tratamiento de pacientes con productos de terapia génica es posible en centros especializados. Varios productos ya han recibido la autorización de comercialización (Tab. 1).
Mensajes para llevarse a casa
- La terapia génica como tratamiento causal de las enfermedades hereditarias monogenéticas se abre paso cada vez más en la clínica.
- Mediante el uso de vectores virales, la terapia génica basada en la adición de genes se ha utilizado con éxito en ensayos clínicos para ciertas enfermedades hematológicas desde el año 2000.
- enfermedades e inmunodeficiencias congénitas.
- La reparación dirigida de genes mediante nucleasas se ha utilizado hasta ahora en pocos estudios para enfermedades oncológicas en las que se quiere desactivar la función de un gen.
- Algunas neoplasias malignas pueden tratarse con células T modificadas genéticamente (células CAR-T).
- Las terapias génicas ya están disponibles actualmente como productos aprobados para 7 enfermedades.
Literatura:
- Ehrhart F, et al: Historia de las enfermedades raras y sus causas genéticas – un enfoque basado en datos. bioRxiv 2020; preprin doi: https://doi.org/10.1101/595819
- Cavazzana-Calvo, M et al: Terapia génica de la enfermedad humana de inmunodeficiencia combinada grave (SCID)-X1. Science 2000; 288: 669-672.
- Kaplitt MG, et al: Safety and Tolerability of Gene Therapy With an Adeno-Associated Virus (AAV) Borne GAD Gene for Parkinson’s Disease: An Open Label, Phase I Trial. Lancet 2007; 369(9579): 2097-2105.
- Todo T: Inmunoterapia activa: terapia vírica oncolítica con VHS-1 Adv Exp Med Biol. 2012; 746: 178-186.
- Brenner AJ, et al: Seguridad y eficacia de VB-111, una terapia génica anticancerosa, en pacientes con glioblastoma recurrente: resultados de un estudio de fase I/II. Neuro Oncol 2020; 22(5): 694-704.
- Palfi S, et al: Seguimiento a largo plazo de un estudio de fase I/II de ProSavin, una terapia génica con vectores lentivirales para la enfermedad de Parkinson. Hum Gene Ther Clin Dev 2018; 29(3): 148-155.
- Hacein-Bey-Abina S, et al: Acontecimiento adverso grave tras una terapia génica exitosa para la inmunodeficiencia combinada grave ligada al cromosoma X. N Engl J Med 2003; 348(3): 255-256.
- Aiuti A, et al.: The Committee for Advanced Therapies’ of the European Medicines Agency Reflection Paper on Management of Clinical Risks Deriving from Insertional Mutagenesis. Human Gene Ther Clin Dev 2013; 24: 47-54.
- Ott MG, et al: Corrección de la enfermedad granulomatosa crónica ligada al cromosoma X mediante terapia génica, aumentada por la activación insercional de MDS1-EVI1, PRDM16 o SETBP1. Nat Med 2006; 12(4): 401-409.
- Siler U, et al.: Combinación exitosa de terapia génica secuencial y alo-HSCT de rescate en dos niños con X-CGD – Importancia del momento oportuno. Curr Gene Ther 2015; 15(4): 416-427.
- Serrao E, et al: Sitios de integración del ADN retroviral: de la investigación básica a las aplicaciones clínicas. AN Crit Rev Biochem Mol Biol 2015; 28: 1-17.
- Manno CS, et al: Transducción exitosa del hígado en la hemofilia mediante AAV-Factor IX y limitaciones impuestas por la respuesta inmunitaria del huésped. Nat Med 2006; 12: 342-347.
- Nathwani AC, et al: Seguridad y eficacia a largo plazo de la terapia génica con factor IX en la hemofilia B. N Engl J Med 2014; 371(21): 1994-2004.
- Aiuti A, et al.: Corrección de ADA-SCID mediante terapia génica de células madre combinada con acondicionamiento no mieloablativo. Science 2002; 296(5577): 2410-2413.
- Mavilio F, et al: Corrección de la epidermólisis bullosa juntural mediante el trasplante de células madre epidérmicas modificadas genéticamente. Nat Med 2006; 12(12): 1397-1402.
- Boztug K, et al: Terapia génica con células madre para el síndrome de Wiskott-Aldrich. N Engl J Med 2010; 363(20): 1918-1927.
- Cartier N, et al.: Terapia génica de células madre hematopoyéticas con un vector lentiviral en la adrenoleucodistrofia ligada al cromosoma X. Science 2009; 326(5954): 818-823.
- Cavazzana-Calvo M, et al.: Independencia de la transfusión y activación del HMGA2 tras la terapia génica de la β-talasemia humana. Naturaleza 2010; 467(7313): 318-322.
- Thompson AA, et al: Terapia génica en pacientes con β-talasemia dependiente de transfusión. N Engl J Med 2018; 378: 147993.
- Aiuti A, et al.: Terapia génica con células madre hematopoyéticas lentivirales en pacientes con síndrome de Wiskott-Aldrich. Science 2013; 341(6148): 1233151.
- De Ravin SS, et al: Terapia génica con células madre hematopoyéticas lentivirales para la inmunodeficiencia combinada grave ligada al cromosoma X. Sci Transl Med 2016; 8(335): 335ra57.
- Mullard A: La EMA da luz verde a una segunda terapia génica. Nat Rev Drug Discov 2016; 15: 299.
- Ribeil JA, et al: Terapia génica en un paciente con anemia falciforme. N Engl J Med 2017; 376(9): 848-855.
- Lwin SM, et al: Resultados de seguridad y eficacia temprana de la terapia génica lentiviral con fibroblastos en la epidermólisis bullosa distrófica recesiva. JCI Insight 2019; 4(11): e126243.
- Biffi A, et al.: La terapia génica con células madre hematopoyéticas lentivirales beneficia a la leucodistrofia metacromática. Science 2013; 341(6148): 1233158.
- Niethammer M, et al: La terapia génica reduce los síntomas de la enfermedad de Parkinson al reorganizar la conectividad funcional del cerebro. Sci Transl Med 2018; 10(469): eaau0713.
- Heiss JD, et al: Ensayo de terapia génica putaminal guiada por resonancia magnética para la enfermedad de Parkinson avanzada. Mov Disord 2019; 34(7): 1073-1078.
- Bainbridge JWB, et al: Efecto de la terapia génica sobre la función visual en la amaurosis congénita de Leber. N Engl J Med 2008; 358(21): 2231-2239.
- Bainbridge JWB, et al: Efecto a largo plazo de la terapia génica en la amaurosis congénita de Leber. N Engl J Med 2015; 372(20): 1887-1897.
- MacLaren RE, et al: Terapia génica retiniana en pacientes con coroideremia: resultados iniciales de un ensayo clínico de fase 1/2. Lancet 2014; 383: 1129-1137.
- Xue K, et al: Efectos beneficiosos sobre la visión en pacientes sometidos a terapia génica retiniana para la coroideremia. Nat Med 2018; 24(10): 1507-1512.
- Rakoczy EP, et al: Gene Therapy With Recombinant Adeno-Associated Vectors for Neovascular Age-Related Macular Degeneration: 1 Year Follow-Up of a Phase 1 Randomised Clinical Trial. Lancet 2015; 386(10011): 2395-2403.
- Feuer WJ, et al: Terapia génica para la neuropatía óptica hereditaria de Leber: resultados iniciales. Oftalmología 2015; 123(3): 558-570.
- Bouquet C, et al: Respuesta inmunitaria e inflamación intraocular en pacientes con neuropatía óptica hereditaria de Leber tratados con inyección intravítrea del virus adeno-asociado recombinante 2 portador del gen ND4: un análisis secundario de un ensayo clínico de fase 1/2. JAMA Oftalmol 2019; 137(4): 399-406.
- Mendell JR, et al: Ensayo de terapia génica con folistatina de fase 1/2a para la distrofia muscular de Becker. Mol Ther 2015; 23(1): 192-201.
- Mendell JR, et al: Terapia de sustitución génica de dosis única para la atrofia muscular espinal. N Engl J Med 2017; 377(18): 1713-1722.
- Corti M, et al: La depleción de células B es protectora frente a la respuesta inmunitaria contra la cápside del VAV: estudio de un caso en humanos. Mol Ther Methods Clin Dev 2014; 1: 14033.
- Calcedo R, et al: Respuestas de células T restringidas a clase I a un péptido polimórfico en un ensayo clínico de terapia génica para la deficiencia de α-1-antitripsina. Proc Natl Acad Sci U S A 2017; 114(7): 1655-1659.
- Mueller C, et al.: Expresión del año y reparación del defecto neutrófilo tras la terapia génica en la deficiencia de alfa-1 antitripsina. Mol Ther 2017; 25(6): 1387-1394.
- Tardieu M, et al: Terapia génica intracerebral en niños con síndrome de mucopolisacaridosis tipo IIIB: un ensayo clínico de fase 1/2 no controlado. Lancet Neurol 2017; 16(9): 712-720.
- Chien YH, et al: Eficacia y seguridad de la terapia génica AAV2 en niños con deficiencia de L-aminoácido aromático descarboxilasa: un ensayo abierto de fase 1/2. Lancet Child Adolesc Health 2017; 1(4): 265-273.
- Kassner U, et al.: Terapia génica en la deficiencia de lipoproteína lipasa: Informe de caso sobre el primer paciente tratado con alipogene tiparvovec en condiciones de práctica diaria. Hum Gene Ther 2018; 29(4): 520-527.
- Kojima K, et al: La terapia génica mejora la función motora y mental de la deficiencia de L-aminoácido aromático descarboxilasa. Cerebro 2019; 142(2): 322-333.
- Porter DL, et al: Células T modificadas por receptores quiméricos de antígenos en la leucemia linfoide crónica. N Engl J Med 2011; 365(8): 725-733.
- Savoldo B, et al: La coestimulación con CD28 mejora la expansión y persistencia de las células T modificadas con receptores de antígenos quiméricos en pacientes con linfoma. J Clin Invest 2011; 121(5): 1822-1826.
- Grupp SA, et al: Células T modificadas con receptores de antígenos quiméricos para la leucemia linfoide aguda. N Engl J Med 2013; 368(16): 1509-1518.
- Yang G, Huang X: Métodos y aplicaciones del sistema CRISPR/Cas para la edición del genoma en células madre. Cell Regen (Lond) 2019; 8(2): 33-41.
PRÁCTICA GP 2020; 15(9): 6-10









