
En el mieloma múltiple, la clínica que conduce al diagnóstico se deriva del daño del órgano final. Los criterios diagnósticos se actualizaron por última vez en 2014. Con diagnósticos bien establecidos y ampliamente disponibles, la enfermedad rara vez causa dificultades de diagnóstico en la actualidad.
Con una incidencia anual de aproximadamente 5-6/100.000 y una cuota del 10%, el mieloma múltiple es una de las neoplasias hematológicas más frecuentes. Con una media de edad de 65-70 años, predominan las personas mayores, y los hombres se ven afectados con más frecuencia que las mujeres, con una proporción de 1,5:1 [1].
Patogénesis
La transformación neoplásica de una célula B del centro germinal en diferenciación a una célula plasmática productora de inmunoglobulinas es el acontecimiento iniciador de la enfermedad para un espectro de enfermedades de células plasmáticas que se manifiestan como gammapatía monoclonal de significado desconocido (GMSI), mieloma latente (MS), mieloma múltiple (MM) o leucemia de células plasmáticas (LCP), dependiendo de la actividad de la enfermedad y de la manifestación clínica [2].
La gammapatía monoclonal de significado incierto es una lesión precursora clonal cuya incidencia aumenta con la edad (se da en aproximadamente el 3% de las personas de >70 años [3]), pero que sólo progresa a mieloma múltiple en aproximadamente el 1% de los casos al año [4,5]. El riesgo exacto de progresión depende del tipo y la concentración de la paraproteína, la proporción de cadenas ligeras libres, el porcentaje de células plasmáticas clonales en la médula ósea y la inmunoparesia [6,7].
Entre medias, el estadio de mieloma asintomático denominado smouldering puede definirse en aproximadamente el 14% de los pacientes, con una tasa de progresión anual del 10% en los cinco primeros años tras el diagnóstico inicial, seguida de un 3% anual en los cinco años siguientes y del 1,5% en los años posteriores [8,9]. Se trata de un estado de enfermedad clínicamente definido entre la GMSI y el mieloma múltiple que incluye una población de pacientes muy heterogénea, entre los que se encuentran pacientes con una progresión de la enfermedad premaligna similar a la GMSI y aquellos con mieloma múltiple agresivo CRAB negativo.
La leucemia de células plasmáticas es la forma más agresiva y leucémica de neoplasia de células plasmáticas, con una incidencia aproximada de 4/10.000.000 [10] es comparativamente rara y puede desarrollarse de forma primaria o secundaria a partir de un mieloma múltiple preexistente (1-4% de todos los pacientes) [11]. Se requiere un porcentaje de células plasmáticas del 20% o una concentración de 2000 células plasmáticas/µl de sangre para el diagnóstico en el recuento sanguíneo diferencial microscópico.
El acontecimiento iniciador de la oncogénesis de las discrasias de células plasmáticas tiene lugar en una fase del desarrollo de las células B que se caracteriza per se por la inestabilidad genética debida a cambios en la clase de isotipo de la molécula de inmunoglobulina y a la hipermutación somática dirigida a la maduración de la afinidad [12].
Citogenéticamente, pueden distinguirse dos cambios principales en el cariotipo, que en el sentido de mutaciones primarias ya están presentes al principio de la oncogénesis en la fase de MGUS. Los cariotipos hiperdiploides, que se observan en casi dos tercios de los casos, se caracterizan por trisomías en cromosomas con número impar (3,5,7,9,11,15,19) y se distinguen del denominado cariotipo no hiperdiploide, que suele estar causado por translocaciones del locus de inmunoglobulina de cadena pesada (IgH) con oncogenes como FGFR-3 y MMSET (t[4;14]), MAF (t[14;16]), CCND1 (t[11;14]) o se caracteriza por ganancias/pérdidas desequilibradas de 1q, 1p, 6q, 8p, 13q, 16q y 17p.
Las alteraciones secundarias incluyen mutaciones en las proteínas RAS (K/N-RAS), mutaciones activadoras en quinasas como PI3K, AKT, BRAF, translocaciones con activación de factores de transcripción como MYC, y deleciones o inactivación de genes supresores de tumores como p53 y RB1 [13].
Al mismo tiempo, el curso de la enfermedad se caracteriza por una creciente heterogeneidad clonal e inestabilidad genómica [14], que posiblemente se ve incrementada por la intervención quimioterapéutica (por ejemplo, agentes alquilantes).
Una vez que se establece un clon de células plasmáticas malignas, se desarrollan daños clínicos en los órganos terminales con una actividad creciente de la enfermedad. Las lesiones óseas osteolíticas debidas a un aumento de la resorción ósea son el resultado de un metabolismo óseo desregulado con un aumento de la actividad de los osteoclastos y una supresión de la de los osteoblastos, mediada por un aumento de la expresión de RANKL (“receptor activador del ligando NF kappa B”), una disminución de la expresión de osteoprotegerina [15] y un entorno de citoquinas de apoyo a los osteoclastos (aumento de MIP-1 alfa, IL6, IL3, etc.). Otra consecuencia de este desequilibrio es la liberación de calcio de la sustancia ósea con la consiguiente hipercalcemia sérica y cambios en la excitabilidad neuromuscular.
La anemia que a menudo conduce al diagnóstico inicial y su aclaración son el resultado del desplazamiento de la hematopoyesis sana por células plasmáticas malignas. Sin embargo, esta explicación se queda corta, ya que en muchos casos se observa una anemia pronunciada que no puede explicarse por una baja infiltración de células plasmáticas demostrada. Aquí parecen desempeñar un papel los cambios en el microentorno de la médula ósea, como la activación de la vía de señalización del TGFβ, que conducen a una concentración reducida de células progenitoras hematopoyéticas en la médula ósea de los pacientes con mieloma [16].
Además de la infiltración directa de células plasmáticas como elemento patogénico, la paraproteína monoclonal secretada por las células plasmáticas desempeña a veces un papel decisivo en la patogénesis. Aunque un síndrome de hiperviscosidad causado por concentraciones elevadas de paraproteína es comparativamente raro y se encuentra entonces sobre todo en enfermedades de mieloma con paraproteína IgM o IgA, lo que puede explicarse por su estructura molecular más compleja con aparición como pentámero (IgM) o dímero (IgA), el daño en los órganos finales causado por el amiloide tóxico de cadena ligera en la patogénesis de la amiloidosis AL, por otra parte, ya se produce con concentraciones bajas de paraproteína. En el mieloma de cadenas ligeras en particular, las cadenas ligeras libres filtrables glomerulares provocan daños tubulares y obstrucción (la denominada nefropatía por colada) debido a su menor tamaño molecular en comparación con la molécula de inmunoglobulina completa.
Clínica
Los síntomas clínicos que en última instancia conducen a la consulta médica y al diagnóstico se derivan de los daños en los órganos finales relacionados con el mieloma descritos en su patogenia. Los dolores óseos debidos a una osteólisis relacionada con el mieloma, una fractura osteolítica patológica, una disminución del rendimiento en presencia de anemia o una tendencia a la infección conducen con frecuencia a una presentación médica. El efecto nefrotóxico de las cadenas ligeras puede provocar insuficiencia renal hasta insuficiencia renal con los correspondientes síntomas urémicos. Más raramente, el mieloma múltiple se manifiesta con arritmias cardiacas, somnolencia u otros síntomas relacionados con la hipercalcemia. El efecto tóxico del amiloide de cadena ligera en la amiloidosis AL puede dar lugar a una sintomatología clínica muy diversa. La insuficiencia cardiaca debida a la deposición cardiaca se encuentra a menudo aquí, junto con la insuficiencia renal, la polineuropatía y otras.
Diagnóstico
Los criterios diagnósticos del mieloma múltiple fueron actualizados por última vez en 2014 por el Grupo Internacional de Trabajo sobre el Mieloma (IMWG) en una actualización de consenso [17].
Los criterios CRAB establecidos (hipercalcemia, insuficiencia renal, anemia, osteólisis) se complementaron con los denominados criterios SLiM (Tab. 1) . El trasfondo de la ampliación de los criterios diagnósticos es la observación de que en el colectivo de pacientes con un mieloma latente que no ha requerido tratamiento previo, determinados parámetros de la enfermedad se asocian a una alta probabilidad de progresión (>80% en un plazo de dos años) se asocian al mieloma múltiple que requiere tratamiento y este grupo de pacientes se beneficia de una intervención terapéutica más temprana [17,18].

Así pues, para el diagnóstico del mieloma múltiple se requiere la detección de ≥10% de células plasmáticas clonales en la biopsia o aspirado de médula ósea. (Fig.1) o se requiere un plasmocitoma óseo o extramedular documentado mediante biopsia, junto con pruebas de uno o más daños en los órganos finales o biomarcadores de malignidad. (Tab.1). La prueba de la clonalidad de las células plasmáticas se consigue mediante la detección por citometría de flujo de una restricción de la cadena ligera citoplasmática. (Fig. 2) o mediante tinción inmunohistoquímica de las cadenas ligeras en una biopsia en sacabocados representativa de la médula ósea.
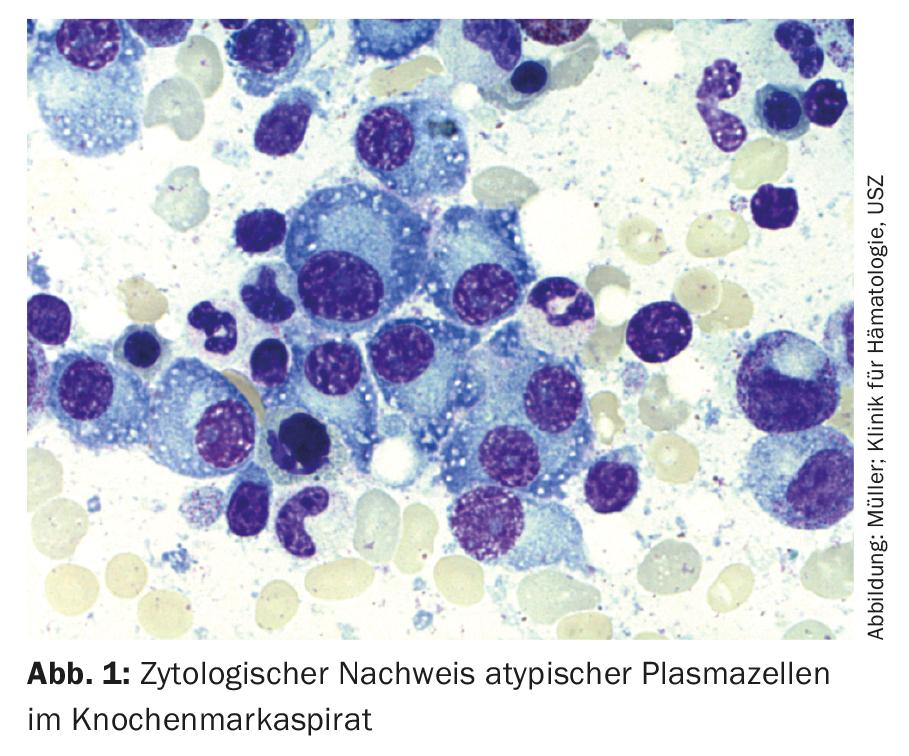
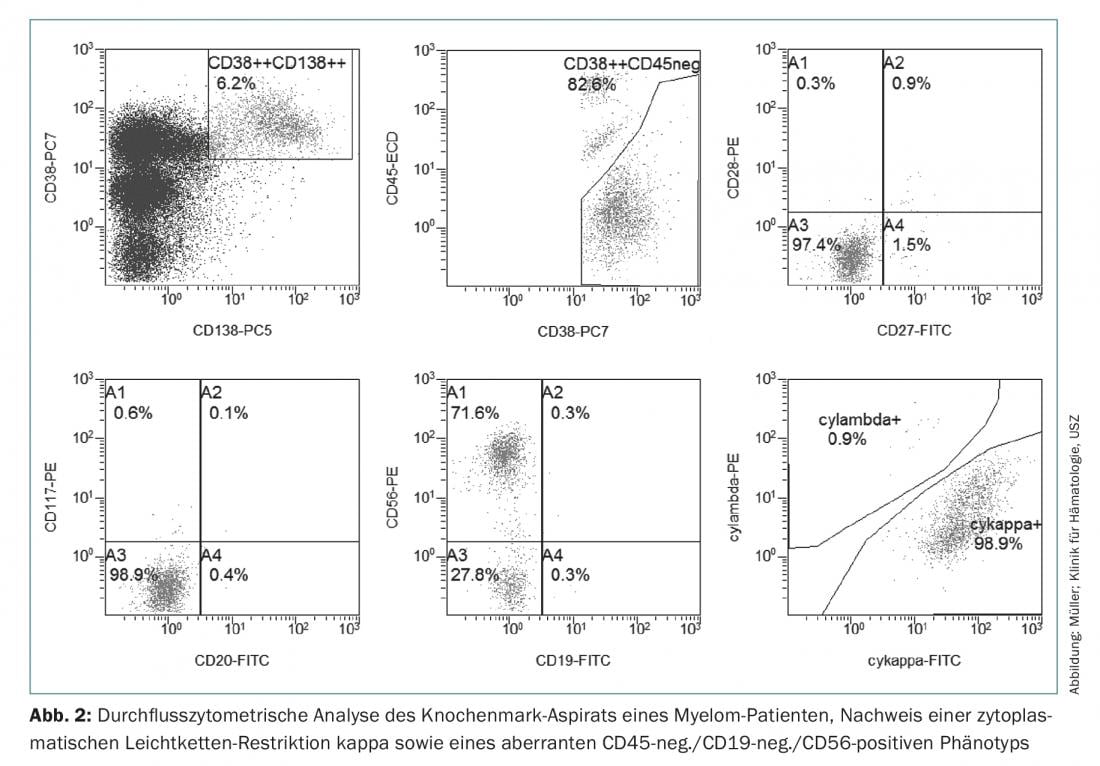
Inmunofenotípicamente, las células del mieloma pueden distinguirse de las células plasmáticas sanas CD38++, CD138+, CD19+, CD45+ y CD56-negativas por la negatividad o expresión reducida de CD45, una pérdida de CD19 y la expresión de marcadores aberrantes como CD56, CD117 o CD28, lo que puede utilizarse en el diagnóstico inicial, pero sobre todo en el diagnóstico de MRD, y a veces también tiene importancia pronóstica [19].
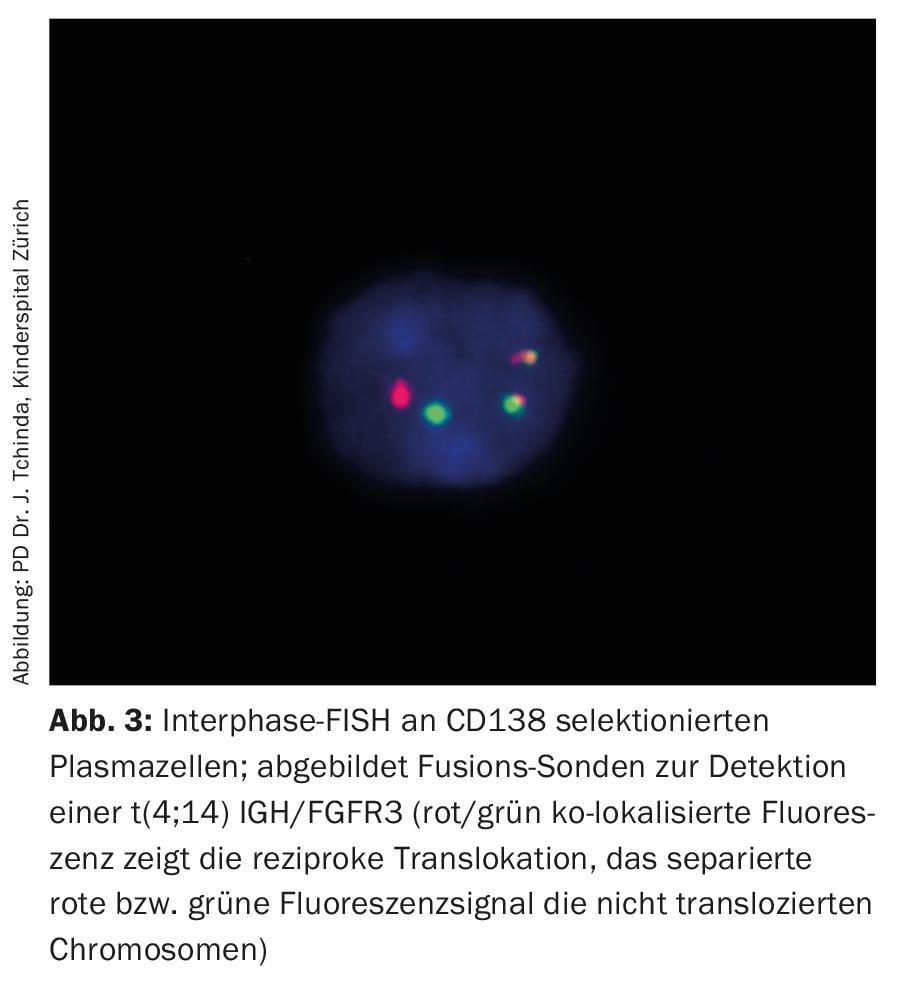
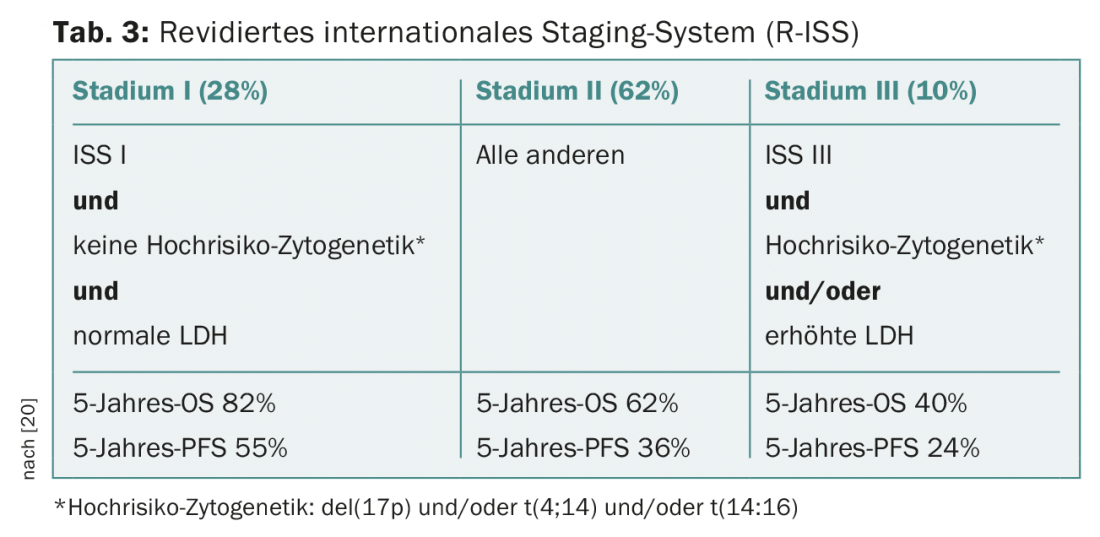
Mientras que el análisis convencional del cariotipo de las metafases detenidas en el mieloma múltiple suele ser poco informativo debido al bajo índice proliferativo y a las difíciles condiciones de cultivo celular, la hibridación fluorescente in situ de los núcleos interfásicos con sondas fluorescentes forma parte de la preparación estándar y, junto con parámetros sustitutos establecidos de la actividad de la enfermedad como la β2-microglobulina, la albúmina y la lactato deshidrogenasa (LDH), permite una estratificación del riesgo a priori. (Fig. 3, Tab. 3) [20].
La tabla 2 ofrece una visión general de los diagnósticos de laboratorio que deben realizarse en el diagnóstico inicial, así como las pruebas de imagen necesarias mediante TC de cuerpo entero a dosis bajas y, opcionalmente, RMN y PET-TC.
Con diagnósticos bien establecidos y ampliamente disponibles, el mieloma múltiple rara vez causa dificultades diagnósticas en la actualidad. Más bien, los retos futuros residen en una subclasificación genética cada vez más determinada de la entidad de la enfermedad con el objetivo de mejorar la estratificación del riesgo y el establecimiento de marcadores predictivos de una respuesta terapéutica.
En el contexto de las opciones de tratamiento cada vez más eficaces con una mejor respuesta y remisiones más profundas, la detección de la enfermedad mínima residual (EMR) mediante citometría de flujo y secuenciación de nueva generación también está desempeñando un papel cada vez más importante en la evaluación de la respuesta y la remisión. Aunque ya se ha demostrado la importancia pronóstica de la ERM para la supervivencia sin progresión (SLP) y la supervivencia global (SG) [21], aún no se han establecido las decisiones terapéuticas clínicas basadas en la ERM, pero son objeto de ensayos clínicos en curso.
Mensajes para llevarse a casa
- La clínica que lleva a la consulta del médico y al diagnóstico se deriva de los daños en los órganos finales relacionados con el mieloma.
- Los criterios diagnósticos del mieloma múltiple se actualizaron por última vez en 2014 como parte de una actualización de consenso.
- Con diagnósticos bien establecidos y ampliamente disponibles, el mieloma múltiple rara vez causa dificultades diagnósticas en la actualidad.
- Más bien, el reto futuro reside en una subclasificación genética cada vez más determinada de la entidad de la enfermedad con el objetivo de mejorar la estratificación del riesgo y el establecimiento de marcadores predictivos de una respuesta terapéutica.
- Se ha demostrado la importancia pronóstica de la enfermedad mínima residual (EMR) para la supervivencia sin progresión (SLP) y la supervivencia global (SG), y las decisiones clínicas de tratamiento basadas en la EMR son objeto de ensayos clínicos actuales.
Literatura:
- Rodríguez-Abreu D, Bordoni A, Zucca E: Epidemiología de las neoplasias hematológicas. Anales de Oncología 2007; 18(Suppl 1): i3-i8.
- Bakkus MH, et al: Pruebas de que los genes VDJ de la cadena pesada de Ig del mieloma múltiple contienen mutaciones somáticas pero no muestran variación intraclonal. Sangre 1992; 80: 2326-2335.
- Kyle RA, et al: Prevalencia de la gammapatía monoclonal de significado indeterminado. N Engl J Med 2006; 354: 1362-1369.
- Landgren O, et al: La gammapatía monoclonal de significado indeterminado (GMSI) precede sistemáticamente al mieloma múltiple: un estudio prospectivo. Sangre 2009; 113: 5412-5417.
- Weiss BM, et al: Una gammapatía monoclonal precede al mieloma múltiple en la mayoría de los pacientes. Sangre 2009; 113: 5418-5422.
- Baldini L, et al.: Papel de diferentes variables hematológicas en la definición del riesgo de transformación maligna en la gammapatía monoclonal. Sangre 1996; 87: 912-918.
- Turesson I, et al: Gammapatía monoclonal de significado indeterminado y riesgo de neoplasias linfoides y mieloides: 728 casos seguidos hasta 30 años en Suecia. Sangre 2014; 123: 338-345.
- Kristinsson SY, Holmberg E, Blimark C: Tratamiento del mieloma latente de alto riesgo. N Engl J Med 2013; 369: 1762-1763.
- Kyle RA, et al: Evolución clínica y pronóstico del mieloma múltiple latente (asintomático). N Engl J Med 2007; 356: 2582-2590.
- Sant M, et al: Incidencia de las neoplasias hematológicas en Europa por subtipo morfológico: resultados del proyecto HAEMACARE. Sangre 2010; 116: 3724-3734.
- Tiedemann RE, et al.: Aberraciones genéticas y supervivencia en la leucemia de células plasmáticas. Leucemia 2008; 22: 1044-1052.
- Seifert M, Scholtysik R, Küppers R: Origen y patogénesis de los linfomas de células B. Métodos Mol Biol 2013; 971: 1-25.
- Kuehl WM, Bergsagel PL: Mieloma múltiple: evolución de los acontecimientos genéticos e interacciones con el huésped. Nat Rev Cancer 2002; 2: 175-187.
- Bolli N, et al.: Heterogeneidad de la evolución genómica y de los perfiles mutacionales en el mieloma múltiple. Nat Commun 2014; 5: 2997.
- Roodman GD: Mecanismos de la metástasis ósea. N Engl J Med 2004; 350: 1655-1664.
- Bruns I, et al.: Desregulación relacionada con el mieloma múltiple de las células madre y progenitoras hematopoyéticas CD34(+) derivadas de la médula ósea. Sangre 2012; 120: 2620-2630.
- Rajkumar SV, et al: Criterios actualizados del Grupo Internacional de Trabajo sobre el Mieloma para el diagnóstico del mieloma múltiple. Lancet Oncol 2014; 15: e538-48.
- Mateos MV, et al: Lenalidomida más dexametasona para el mieloma múltiple latente de alto riesgo. N Engl J Med 2013; 369: 438-447.
- Mateo G, et al: Valor pronóstico del inmunofenotipo en el mieloma múltiple: un estudio de los grupos de estudio cooperativo PETHEMA/GEM en pacientes tratados uniformemente con terapia de dosis alta. Revista de Oncología Clínica 2008; 26: 2737-2744.
- Palumbo A, et al: Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol 2015 Sep 10; 33(26): 2863-2869.
- Paiva B, van Dongen JJM, Orfao A: Nuevos criterios para la evaluación de la respuesta: papel de la enfermedad mínima residual en el mieloma múltiple. Sangre 2015; 125: 3059-3068.
- Moreau P, et al: Mieloma múltiple: ¿Guías de práctica clínica de la ESMO para el diagnóstico, el tratamiento y el seguimiento? Ann Oncol 2017 Jul 1; 28(suppl_4): iv52-iv61.
InFo ONCOLOGÍA Y HEMATOLOGÍA 2017; 5(5): 7-10









