La neoplasia mieloproliferativa de células madre hematopoyéticas está causada predominantemente por mutaciones somáticas del gen JAK2 y la hematopoyesis autónoma clonal resultante. El riesgo de mielofibrosis o de transformación leucémica son relevantes desde el punto de vista pronóstico.
La policitemia vera (PV) está clasificada actualmente por la Organización Mundial de la Salud (OMS) en la categoría principal de las neoplasias mieloproliferativas (NMP) [1,2]. Según los conocimientos actuales, la PV es una enfermedad de las células madre o progenitoras hematopoyéticas, causada predominantemente por mutaciones somáticas en el gen JAK2, con la consiguiente mieloproliferación clonal [3]. Se han descrito grupos familiares poco frecuentes, con mutaciones de la línea germinal en el receptor de la eritropoyetina (EPOR) [4]. La incidencia de la PV en Europa es del 0,4-2,8% de la población al año. La edad media de aparición es de 60-65 años, y mujeres y hombres se ven afectados en un número aproximadamente igual [5,6]. La mediana de supervivencia es de 14 años en los pacientes con PV de más edad (>60 años) y de 24 años en los menores de 60 años [7].
Clínica
En el cuadro sanguíneo, la enfermedad se manifiesta por un aumento de la producción de eritrocitos (eritrocitosis) independiente de los mecanismos fisiológicos de regulación. La leucocitosis y la trombocitosis debidas al aumento de la megacariopoyesis suelen acompañar a la enfermedad. En el curso de la enfermedad, los cambios fibróticos progresivos en el sentido de la mielofibrosis conducen a una denominada “fase gastada” con hematopoyesis reducida en hasta un 10% de todos los pacientes con PV [8]. La transformación leucémica se diagnostica hasta en un 5% de los casos de PV tras 20 años de progresión de la enfermedad [7,9]. Los factores de riesgo pronóstico de mielofibrosis, transformación leucémica y reducción de la supervivencia global son la edad avanzada del paciente, la leucocitosis, la trombocitosis y un cariotipo anormal [10].
Además, la aparición de episodios tromboembólicos venosos y arteriales en el 20-50% de todos los pacientes con PV tiene una relevancia pronóstica decisiva. Fisiopatológicamente, se basan en el aumento de la viscosidad sanguínea y los cambios reológicos resultantes, así como en estímulos inflamatorios, procoagulantes y microvasculares [11,12]. Se distinguen dos grupos de riesgo de trombosis recurrente: Alto riesgo en pacientes >60 años y antecedentes positivos de acontecimientos trombóticos y un grupo de bajo riesgo en ausencia de ambos factores [13]. La hipertensión arterial es un factor de riesgo independiente para la aparición de trombos arteriales.
Clínicamente, la PV se manifiesta principalmente por fatiga (hasta en un 84,9%) [14] y prurito (en cerca del 40%) [15]. Otros síntomas clínicos son enrojecimiento de la piel de la cara, piel y mucosas azuladas, presión en la cabeza, dolor de cabeza e hipertensión. Los trastornos microcirculatorios suelen dar lugar a síntomas clínicos característicos (por ejemplo, alteraciones visuales, parestesias, eritromelalgias, molestias pectanginales) [16]. La trombosis venosa abdominal y la trombosis venosa sinusal son otras complicaciones con consecuencias graves y a menudo mortales [17,18]. Paradójicamente, en casos raros con trombocitosis extrema (>1000×109/L) también hay una mayor tendencia a las hemorragias. La causa es una reducción del factor Von Willebrand (FVW) en el sentido de un síndrome de consumo adquirido. Desde el punto de vista patobiológico, tanto el aumento de la actividad de ADAMTS13 como el aumento de la sensibilidad plaquetaria y la consiguiente activación inadecuada de las plaquetas desempeñan un papel importante [19,20]. La esplenomegalia se encuentra en aproximadamente un tercio de los afectados y puede asociarse a dolor e infarto.
Patobiología de la PV
La PV se caracteriza por una mieloproliferación clonal debida a cambios patológicos en las células madre hematológicas [3]. Casi todos los pacientes con PV presentan una mutación de la Janus quinasa 2 (JAK2), que se localiza en el locus génico 9p24, y el 96% de todos los casos tienen una mutación somática activadora en el exón 14 (JAK2V617F) [21] y con menor frecuencia (alrededor del 3%) en el exón 12 [22]. Hasta ahora, no se han demostrado diferencias significativas en el curso clínico en función de la mutación respectiva [23].
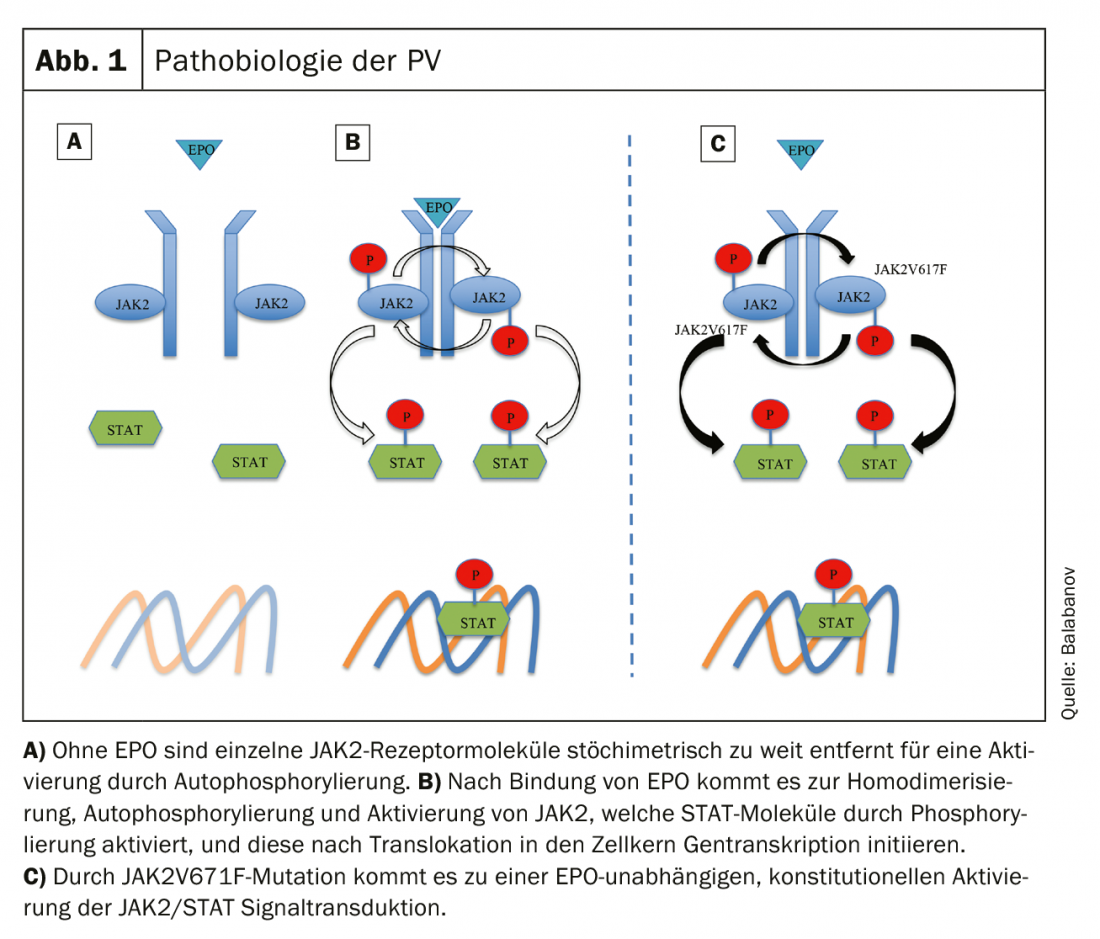
En condiciones fisiológicas, la unión de la eritropoyetina (EPO) al receptor de la EPO (EPOR) provoca un cambio conformacional (homodimerización del receptor) con la consiguiente autofosforilación y activación de la JAK. Las moléculas JAK activadas fosforilan y posteriormente activan las moléculas STAT (transductor de señales y activador de la transcripción) que, tras su translocación al núcleo celular, desencadenan señales de crecimiento hematopoyético como factores de transcripción [24]. La mutación JAK2V617F activadora da lugar a una activación JAK/STAT constitucional independiente de la EPO y a una proliferación celular descontrolada (Fig. 1) [25]. Se han descrito otras alteraciones genéticas como la desregulación del apotosinhibidor Bcl-x o del factor de transcripción NF-E2, pero parecen ser consecuencias secundarias de una mutación JAK2 [26].
Las mutaciones JAK2V617F son más frecuentes en pacientes de edad avanzada y se asocian a panmielocitosis, mielofibrosis y síntomas clínicos de prurito [22,23]. No se pudo demostrar una correlación con la supervivencia global o el riesgo de transformación [3]. La frecuencia de alelos mutantes tampoco parece influir en la supervivencia ni en la frecuencia de transformación leucémica. En un estudio de la Clínica Mayo, la secuenciación profunda dirigida mostró que más de la mitad (53%) de los pacientes con PV examinados presentaban mutaciones adicionales además de la JAK2 [28]. Las mutaciones más frecuentes se detectaron en TET2 (22%), ASXL1 (12%), SH2B3 (9%) (se encontraron otras mutaciones en SRSF2, IDH2, TP53). Especialmente las mutaciones en ASXL1, SRSF2 y DHI2 mostraron un efecto pronóstico independiente de otros predictores (mediana de supervivencia 7,7 años con vs. 16,9 años sin mutaciones). Se encuentra un cariotipo anormal en el 15% de los pacientes en el diagnóstico inicial, siendo las alteraciones citogenéticas más comunes detectadas -Y, +8, +9, del(20q) y 1q+ [27].
Diagnóstico
Una historia clínica y un examen físico detallados constituyen la base del diagnóstico. El cuestionario de evaluación de los síntomas de las neoplasias mieloproliferativas (MPN-SAF) proporciona una evaluación estructurada y completa de los síntomas relevantes [29]. En particular, debido a la relevancia pronóstica y terapéutica, debe prestarse atención a los acontecimientos trombóticos en los antecedentes, los estigmas de la trombosis, la hipertensión y la edad del paciente. Se recomienda la ecografía abdominal para evaluar la esplenomegalia.
Además de un recuento sanguíneo diferencial y del estado de coagulación, debe determinarse la concentración de EPO en el laboratorio. El diagnóstico definitivo sólo puede hacerse a partir de una biopsia de médula ósea.
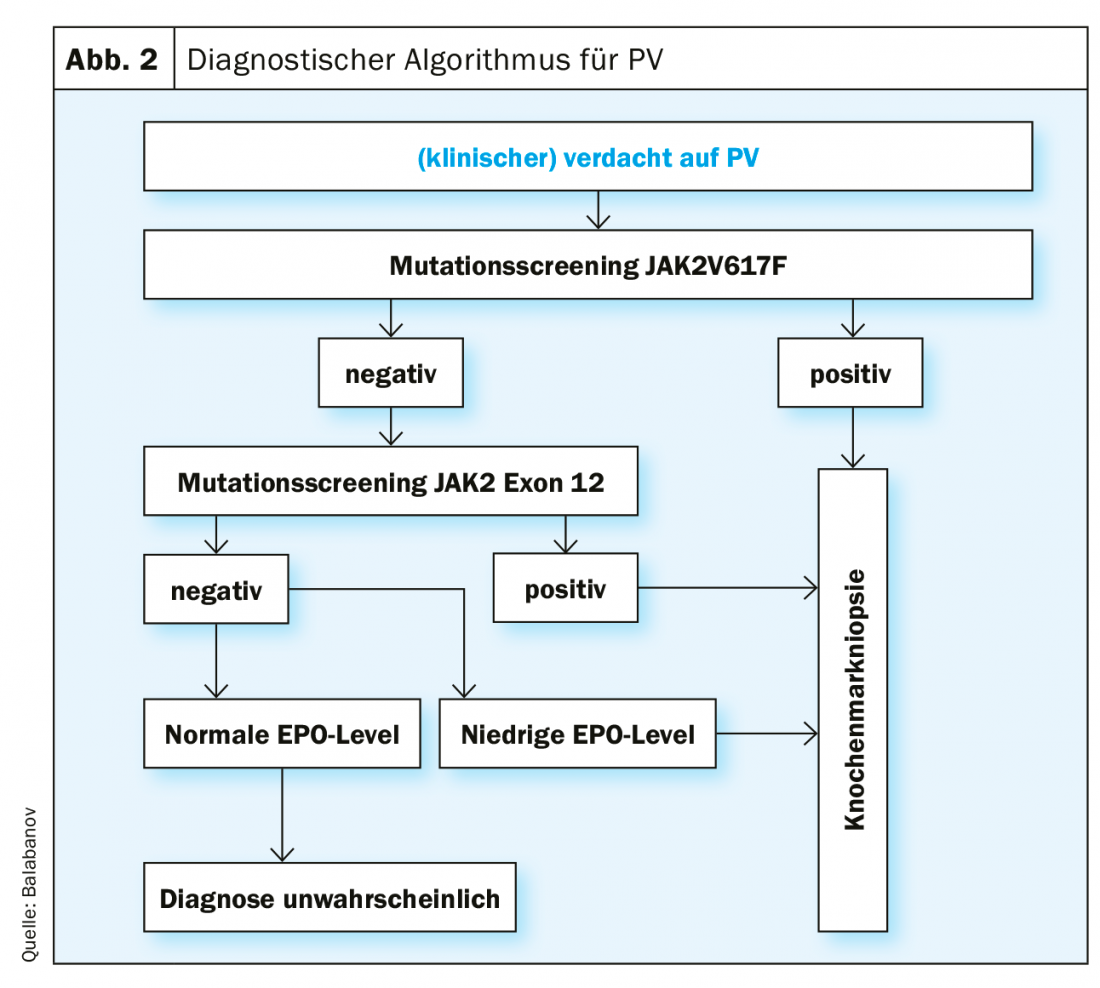
Los criterios diagnósticos según la clasificación actual (2016) de la OMS se muestran en la tabla 1. Para hacer un diagnóstico de PV, deben cumplirse todos los criterios mayores o dos criterios mayores y el criterio menor. La detección de una mutación JAK2V617F es extremadamente fiable, con una sensibilidad de la prueba del 97% y una especificidad de casi el 100%. Si el estado de la mutación JAK2V617F es negativo pero el nivel sérico de EPO es bajo, está indicado un análisis adicional de la mutación en el exón 12 del gen JAK2. Debido a la relevancia pronóstica tanto para la supervivencia global como para la supervivencia libre de leucemia, debe realizarse una determinación del cariotipo si es necesario. Además, resulta útil un análisis de mutaciones mediante secuenciación de próxima generación (NGS), ya que las mutaciones en ASXL1, SRSF2 e IDH2 tienen relevancia pronóstica. Sin embargo, la NGS aún no es una investigación estándar para todos los pacientes con PV en el momento del diagnóstico. Sin embargo, un estudio histórico publicado recientemente ha demostrado que la integración global de variables genómicas y clínicas permite una estratificación personalizada del riesgo con implicaciones terapéuticas [30] (https://cancer.sanger.ac.uk/mpn-multistage).

En la figura 2 se muestra un algoritmo para el procedimiento diagnóstico de la sospecha de PV, basado en los criterios diagnósticos actuales de la OMS. Las pruebas funcionales especiales del FvW están indicadas para aclarar una tendencia hemorrágica aumentada (por ejemplo, la actividad del cofactor de ristocitina).
Terapia
Con una tasa de supervivencia a 10 años superior al 75% y un riesgo relativamente bajo de mielofibrosis (<10%) y de transformación maligna en el sentido de transformación leucémica (<5%) [8], el objetivo principal del tratamiento de la PV es prevenir las complicaciones trombohemorrágicas y aliviar los síntomas descritos anteriormente.
Pacientes de bajo riesgo (edad <60 años, antecedentes negativos de acontecimientos trombóticos) : Recomendamos un tratamiento flebotómico constante, con el objetivo de alcanzar un hematocrito <45%, ya que este valor objetivo es superior a valores superiores (45-50%) en términos de supervivencia libre de eventos [31]. Por otro lado, aún no se ha documentado un efecto positivo sobre los síntomas de la enfermedad [34]. Además, se recomienda la terapia antiagregante con aspirina (AAS) para prevenir los trombos venosos y arteriales [31–33]. El tratamiento con AAS parece influir positivamente en los síntomas asociados al reducir las alteraciones microvasculares [35]. En caso de síntomas resistentes a la aspirina, puede considerarse la administración dos veces al día o, alternativamente, el uso de clopidogrel solo o en combinación con AAS [36, 37]. Si es necesario, está indicada una prueba de la función plaquetaria (agregometría plaquetaria, PFA-100) para distinguir los defectos congénitos de las causas iatrogénicas [38]. Se aconseja precaución en el uso de aspirina en vista del posible aumento de la tendencia hemorrágica en pacientes PV con trombocitosis extrema concomitante (>1000×109/L).
Pacientes de alto riesgo (edad >60 años, antecedentes positivos de eventos trombóticos): Además de las medidas terapéuticas mencionadas, recomendamos el tratamiento citorreductor en este grupo de pacientes, siendo el objetivo principal la normalización del hematocrito. Sin embargo, también debe perseguirse una reducción de la trombocitosis y la leucocitosis que a menudo la acompañan. Las recomendaciones actuales de sustancias para la terapia de primera línea según la European LeukemiaNET (ENL) son hidroxiurea e interferón α (INFα) (forma pegilada) [39]. El fracaso del tratamiento con hidroxiurea (tab. 2 ) en el sentido de resistencia o intolerancia se produce en aproximadamente el 24% de los pacientes [7]. El busulfán se ha utilizado históricamente con buen éxito en la terapia de segunda línea. Hoy en día, su uso es esencialmente obsoleto debido a la sospecha de un potencial leucemógeno, por lo que no se dispone de datos sólidos con respecto al aumento de la aparición de leucemias bajo busulfán [40].

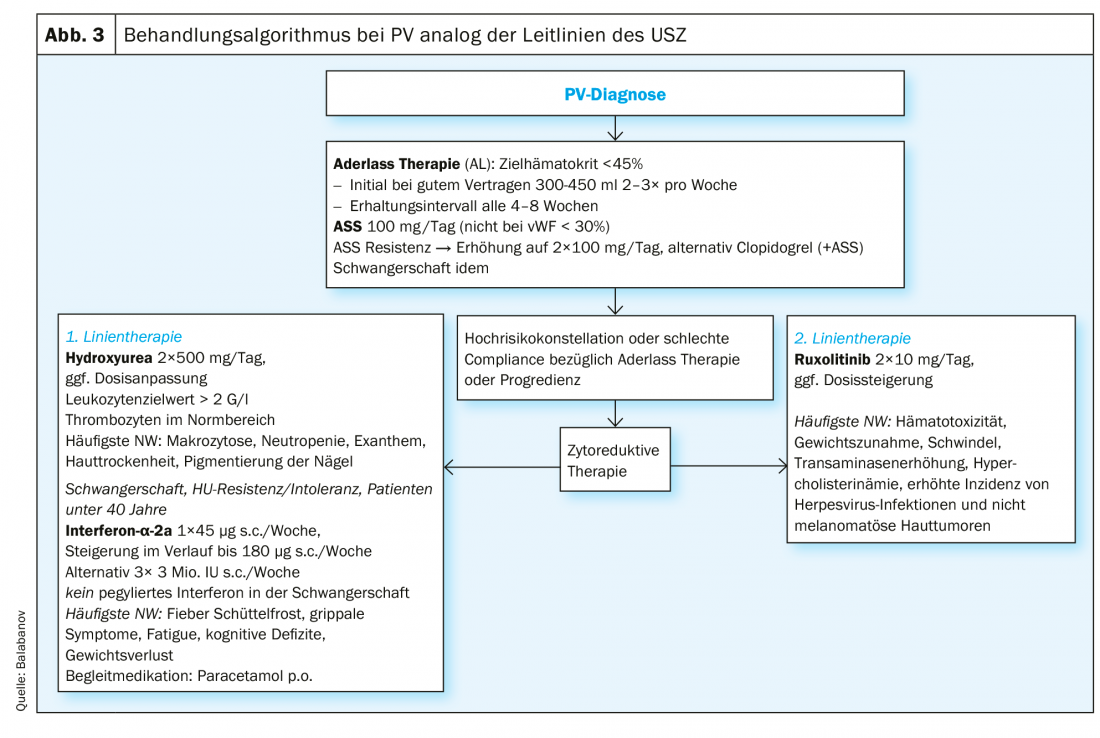
El inhibidor de JAK2 ruxolitinib está aprobado como terapia de segunda línea para la PV y mostró resultados positivos en términos de mieloproliferación, necesidad de flebotomía y síntomas asociados a la PV como fatiga y prurito con buena tolerabilidad [41–44]. Para el prurito refractario, se ha descrito el beneficio del uso fuera de indicación de ISRS [45], INFα [46] y fototerapia [47]. En la figura 3 se muestra un resumen del algoritmo de tratamiento del Hospital Universitario de Zúrich (USZ). El trasplante alogénico de médula ósea o el trasplante de células madre de sangre periférica deben considerarse como un enfoque curativo sólo en casos excepcionales en pacientes jóvenes con un curso rico en complicaciones o una progresión de la enfermedad que no pueda controlarse con otras medidas [48]. Debido al aumento del riesgo cardiovascular, debe animarse a todos los pacientes a reducir el riesgo cardiovascular en términos de prevención primaria (ejercicio, control del peso, dieta).
Embarazo: la PV no es una contraindicación para el embarazo, pero siempre se consideran embarazos de alto riesgo, con una mayor tasa de abortos espontáneos en comparación con la población normal [49]. En principio, recomendamos la atención en un centro especializado con los conocimientos hematológicos adecuados. Las recomendaciones relativas a la terapia de flebotomía y a la terapia antiagregante son esencialmente las mismas que las mencionadas anteriormente. La aspirina parece reducir la tasa de abortos puntuales y las complicaciones del embarazo [50]. Debido a la falta de pruebas de teratogenicidad o de influencia en la tasa de nacidos vivos [51] con terapia de IFNα es posible en general y se recomienda especialmente en embarazos de alto riesgo [52].
Mensajes para llevarse a casa
- La policitemia vera es una neoplasia mieloproliferativa de las células madre hematopoyéticas desencadenada predominantemente por mutaciones somáticas del gen JAK2 y que da lugar a una hematopoyesis clonal autónoma.
- Clínicamente, destacan los síntomas y complicaciones mirco y macrovasculares en el contexto de la eritrocitosis y la trombocitosis acompañante frecuente. De mayor relevancia pronóstica es el riesgo de mielofibrosis y transformación leucémica.
- Además de los parámetros clínicos y de laboratorio, el algoritmo de diagnóstico implica principalmente la detección de una mutación JAK2 en la médula ósea, aunque otras aberraciones genéticas adquieren cada vez más importancia.
- Desde el punto de vista terapéutico, los pacientes se dividen en grupos de riesgo. Mientras que la terapia flebotómica y los inhibidores de la agregación plaquetaria se utilizan independientemente del riesgo, la terapia citorreductora está indicada principalmente en pacientes de alto riesgo.
- Los embarazos en pacientes con PV se consideran siempre embarazos de alto riesgo y deben gestionarse de forma multidisciplinar en centros especializados.
Literatura:
- Arber DA, et al: La revisión de 2016 de la clasificación de la Organización Mundial de la Salud de las neoplasias mieloides y la leucemia aguda. Sangre, 2016. 127(20): 2391-2405.
- Barbui T, et al: La revisión de 2016 de la clasificación de la OMS de las neoplasias mieloproliferativas: Avances clínicos y moleculares. Blood Rev, 2016. 30(6): 453-459.
- Jamieson CH, et al.: La mutación JAK2 V617F se produce en las células madre hematopoyéticas en la policitemia vera y predispone hacia la diferenciación eritroide. Proc Natl Acad Sci U S A, 2006. 103(16): 6224-6229.
- Bento C, et al: Bases genéticas de la eritrocitosis congénita: actualización de mutaciones y bases de datos en línea. Hum Mutat, 2014. 35(1): 15-26.
- Policitemia vera: la historia natural de 1213 pacientes seguidos durante 20 años. Gruppo Italiano Studio Policitemia. Ann Intern Med, 1995. 123(9): 656-664.
- Moulard O, et al: Epidemiología de la mielofibrosis, la trombocitemia esencial y la policitemia vera en la Unión Europea. Eur J Haematol, 2014. 92(4): 289-297.
- Tefferi A, et al.: Supervivencia a largo plazo y transformación blástica en la trombocitemia esencial, la policitemia vera y la mielofibrosis anotadas molecularmente. Sangre, 2014. 124(16): 2507-2513; quiz 2615.
- Crisa E, et al.: Estudio retrospectivo en 226 pacientes con policitemia vera: impacto del valor medio del hematocrito en los resultados clínicos y mejora de la supervivencia con profilaxis antitrombótica y fármacos no alquilantes. Ann Hematol, 2010. 89(7): 691-699.
- Barbui T., et al: La supervivencia y la progresión de la enfermedad en la trombocitemia esencial se ven influidas significativamente por un diagnóstico morfológico preciso: un estudio internacional. J Clin Oncol, 2011. 29(23): 3179-3184.
- Tefferi A, et al: Supervivencia y pronóstico entre 1545 pacientes con policitemia vera contemporánea: un estudio internacional. Leucemia, 2013. 27(9): 1874-1881.
- Koschmieder S, et al.: Neoplasias mieloproliferativas e inflamación: si hay que dirigirse al clon maligno, al proceso inflamatorio o a ambos. Leucemia, 2016. 30(5): 1018-1024.
- Kroll MH, Michaelis LC, Verstovsek S: Mecanismos de la trombogénesis en la policitemia vera. Blood Rev, 2015. 29(4): 215-221.
- Finazzi G, Barbui T: Evidencias y experiencia en el tratamiento de la policitemia vera y la trombocitemia esencial. Leucemia, 2008. 22(8): 1494-1502.
- Mesa RA, et al: The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cáncer, 2007. 109(1): 68-76.
- Saini KS, Patnaik MM, Tefferi A: Prurito asociado a la policitemia vera y su tratamiento. Eur J Clin Invest, 2010. 40(9): 828-834.
- Michiels JJ: Eritromelalgia y complicaciones vasculares en la policitemia vera. Semin Thromb Hemost, 1997. 23(5): 441-454.
- De Stefano V, et al.: Trombosis de la vena esplácnica y neoplasias mieloproliferativas: diagnóstico molecular y tratamiento a largo plazo. Thromb Haemost, 2016. 115(2): 240-249.
- Dentali F, et al: Trombosis venosa cerebral y neoplasias mieloproliferativas: resultados de dos grandes bases de datos. Thromb Res, 2014. 134(1): 41-43.
- Elliott MA, Tefferi A: Trombosis y hemorragia en la policitemia vera y la trombocitemia esencial. Br J Haematol, 2005. 128(3): 275-290.
- Michiels JJ, et al.: La paradoja de la activación plaquetaria y el deterioro de la función: las interacciones plaqueta-factor von Willebrand y la etiología de las manifestaciones trombóticas y hemorrágicas en la trombocitemia esencial y la policitemia vera. Semin Thromb Hemost, 2006. 32(6): 589-604.
- Pardanani A, et al: Prevalencia y correlatos clinicopatológicos de las mutaciones del exón 12 de JAK2 en la policitemia vera JAK2V617F-negativa. Leucemia, 2007. 21(9): 1960-1963.
- Vannucchi AM, et al.: Correlatos clínicos de la presencia de JAK2V617F o de la carga alélica en las neoplasias mieloproliferativas: una reevaluación crítica. Leucemia, 2008. 22(7): 1299-1307.
- Passamonti F, et al.: Un estudio prospectivo de 338 pacientes con policitemia vera: el impacto de la carga del alelo JAK2 (V617F) y la leucocitosis en la transformación fibrótica o leucémica de la enfermedad y las complicaciones vasculares. Leucemia, 2010. 24(9): 1574-1579.
- Ward AC, Touw I, Yoshimura A: La vía Jak-Stat en la hematopoyesis normal y perturbada. Sangre, 2000. 95(1): 19-29.
- James C, et al: Una mutación clonal única de JAK2 que conduce a una señalización constitutiva causa la policitemia vera. Nature, 2005. 434(7037): 1144-1148.
- Baxter EJ, et al: Mutación adquirida de la tirosina quinasa JAK2 en los trastornos mieloproliferativos humanos. Lancet, 2005. 365(9464): 1054-1061.
- Gangat N, et al.: Estudios citogenéticos en el momento del diagnóstico en la policitemia vera: correlatos clínicos y de carga alélica JAK2V617F. Eur J Haematol, 2008. 80(3): 197-200.
- Tefferi A, et al: Secuenciación profunda dirigida en la policitemia vera y la trombocitemia esencial. Blood Adv, 2016. 1(1): 21-30.
- Scherber R, et al: The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Sangre, 2011. 118(2): 401-408.
- Grinfeld J, et al: Clasificación y pronóstico personalizado en las neoplasias mieloproliferativas. N Engl J Med, 2018. 379(15): 1416-1430.
- Marchioli R, et al: Acontecimientos cardiovasculares e intensidad del tratamiento en la policitemia vera. N Engl J Med, 2013. 368(1): 22-33.
- Landolfi R, et al: Eficacia y seguridad de las dosis bajas de aspirina en la policitemia vera. N Engl J Med, 2004. 350(2): 114-124.
- Alvarez-Larran A, et al.: Observación frente a terapia antiplaquetaria como profilaxis primaria de la trombosis en la trombocitemia esencial de bajo riesgo. Sangre, 2010. 116(8): 1205-1210; quiz 1387.
- Grunwald MR, et al: Características clínicas y de la enfermedad de REVEAL en el momento de la inscripción (línea de base): Estudio observacional prospectivo de pacientes con policitemia vera en Estados Unidos. Clin Lymphoma Myeloma Leuk, 2018. 18(12): 788-795 e2.
- Michiels JJ, et al: Eritromelalgia mediada por plaquetas, cerebral, ocular y coronaria.
InFo ONCOLOGÍA Y HEMATOLOGÍA 2019; 7(2-3): 12-15.









