Los gliomas de grado II tienden a subestimar su malignidad en la fase inicial. No existe una norma para el tratamiento. Además de la espera, cada vez se adoptan más tácticas quirúrgicas agresivas. Sin embargo, con esto tampoco se consigue ninguna cura.
Los gliomas poco malignos de grado II de la OMS suelen infiltrar la corteza y, por lo tanto, provocan crisis epilépticas en el 60-80% de los casos, a menudo como manifestación inicial [1,2]. El lóbulo más grande del cerebro, el frontal, es el que se ve afectado con más frecuencia. Se producen cambios de personalidad, alteraciones del impulso y del estado de ánimo, pero también ataxia frontal. Si está afectada la región fronto-precentral a postcentral, se producen crisis motoras focales persistentes con tendencia a la generalización, pero también hemiparesia espástica y alteraciones sensoriales. Si está afectado el lóbulo temporal, predominan las crisis parciales complejas e incluso los trastornos del habla. Los trastornos del sistema visual son raros, siendo la causa más probable la hemianopsia homónima cuando está afectado el tractus opticus. Si el tronco encefálico está afectado, se producen trastornos neurológicos complejos de las vías largas y de la función de los nervios craneales, hasta la parálisis bulbar con trastornos de la deglución y aspiración. Si se ven afectados el tálamo y los ganglios basales, se producen trastornos extrapiramidales o trastornos neurológicos. Las fluctuaciones en la vigilancia hasta el punto de la demencia pasan a primer plano. A menudo, sin embargo, la personalidad se conserva bien durante mucho tiempo, incluso en el caso de procesos muy grandes, porque las células tumorales crecen difusamente a través del tejido cerebral sano sin destruirlo. El lento desplazamiento de las zonas funcionales por el tejido tumoral permite un desplazamiento de la función amenazada hacia las zonas vecinas o hacia el lado opuesto (plasticidad), lo que es reconocible en la resonancia magnética funcional.
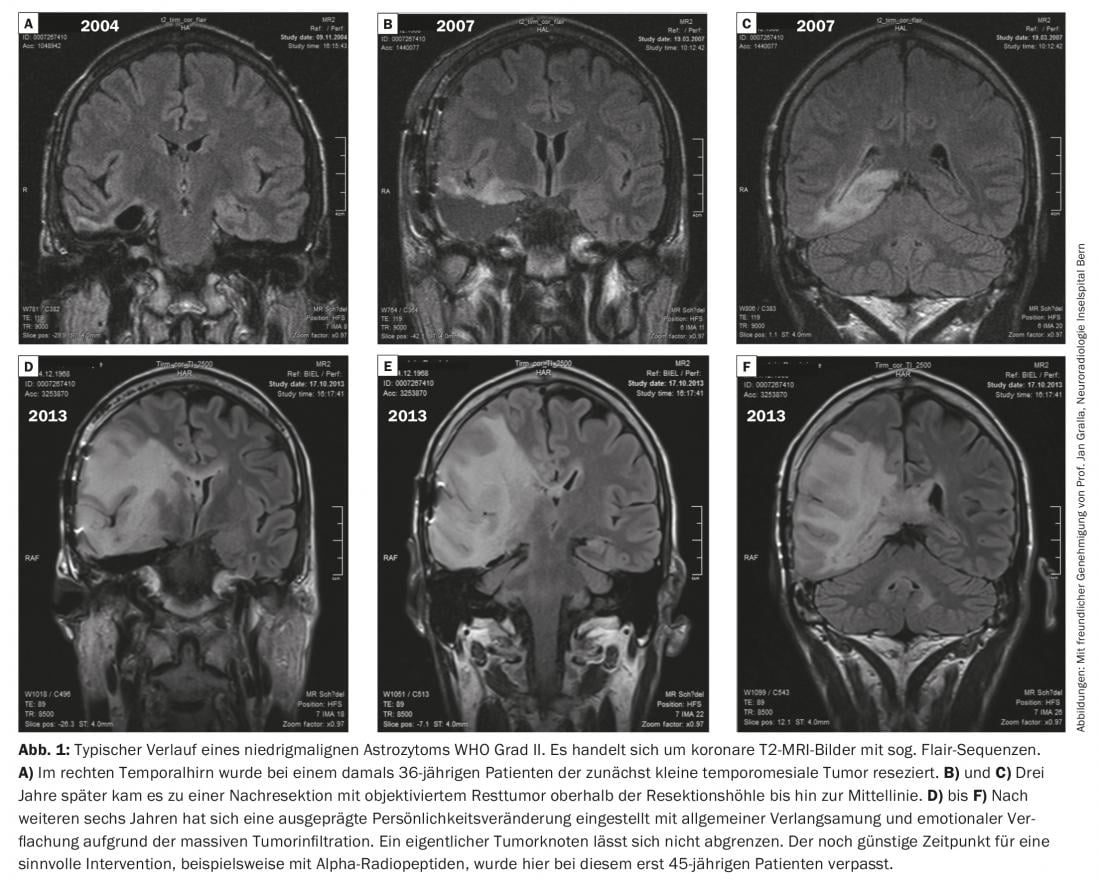
La figura 1 muestra la evolución típica de un astrocitoma poco maligno de grado II de la OMS.
Gliomas: ¿Manifestación de la tasa de mutación espontánea?
En raras ocasiones (<5%), los tumores cerebrales aparecen de forma sindrómica en cánceres familiares, por ejemplo en el síndrome de Turcot con defecto del gen “mismatch repair” o en el síndrome de Li-Fraumeni con mutación p53 [3–5]. Aún más raros son los astrocitomas benignos de células gigantes que se manifiestan en la infancia y esporádicamente en adultos como resultado de una mutación congénita de los genes de la esclerosis tuberosa TSC1 y TSC2, que corregulan el complejo mTOR del metabolismo energético. El defecto genético más común asociado a las enfermedades neurológicas es la mutación del gen de la neurofibromatosis tipo 1, con una frecuencia de 1:3000. Se trata de microdeleciones intragénicas, que en la mitad de los casos no se heredan, sino que se producen espontáneamente. El locus del gen NF1 en el cromosoma 17q11.2 es relativamente inestable. El fenotipo Nf1 incluye los gliomas ópticos benignos. Las deleciones de Nf1 se han detectado en subtipos de glioblastoma y contribuyen a la génesis del glioma en combinación con otras mutaciones. Las mutaciones de los genes de la isocitrato deshidrogenasa IDH1 e IDH2 se encuentran con frecuencia en los astrocitomas de grado II y en el glioblastoma secundario de mejor pronóstico. Al igual que la codeleción 1p-19q en el oligodendroglioma, la mutación IDH marca un tipo de origen biológico diferente con un mejor pronóstico.
La tasa de mutación espontánea es de 1:100.000 por división celular. Un organismo pasa por unas1014 divisiones celulares hasta la diferenciación completa. A lo largo de la vida, se producen innumerables errores de lectura en cada célula durante la división celular -también en la reserva de células madre- que, en su mayoría, son corregidos inmediatamente por mecanismos de reparación intrínsecos a la célula. No obstante, siempre hay mutaciones que no se reconocen o detectan. no se repara, lo que en raras ocasiones contribuye al desarrollo de tumores. Los gliomas pertenecen a las llamadas “enfermedades huérfanas”, a las enfermedades muy raras, con una incidencia inferior a cinco casos por cada 10.000 personas al año.
Clasificación genética molecular
Las pruebas genéticas recientemente desarrolladas que escanean todo el genoma permiten un pronóstico bastante preciso de todos los tipos de glioma y permiten asignar correctamente los casos histológicamente poco claros [6]. Se escanea todo el genoma en busca de patrones de metilación patológicos. La expresión de muchos genes que controlan la proliferación de las células tumorales y promueven la apoptosis está regulada por las llamadas islas CpG en la región promotora de los genes, que pueden desactivarse por metilación, por ejemplo, en la diferenciación de órganos, pero también en las células cancerosas. Otro tipo de inactivación genética es la deleción de largo alcance de los brazos cromosómicos, transformando una sección heterocigótica en una sección monocigótica, a menudo con reduplicación. Ahora se puede crear un denominado alelotipo sobre todos los cromosomas para registrar la pérdida de heterocigosidad. Por ejemplo, las monosomías 1p y 19q son patognomónicas de los oligodendrogliomas de crecimiento lento. Los oligodendrogliomas de tipo salvaje con heterocigosidad 1p/19q se comportan de forma mucho más agresiva. En los glioblastomas suelen encontrarse la monosomía del cromosoma 10 y la trisomía del cromosoma 7 con amplificación del EGFR. El análisis combinado del alelotipo y el metiloma permite una clasificación muy precisa de todos los gliomas, especialmente en los casos en los que la histología plantea dudas. Si en los estudios sobre el glioblastoma se observan los denominados supervivientes a largo plazo, el diagnóstico histológico debe comprobarse con pruebas moleculares, ya que la tasa de error histológico en los estudios a gran escala es de hasta el 7% [7].
Retraso en la reducción de la masa al manifestarse los síntomas
Dado que la mayoría de los pacientes con glioma de grado II se encuentran en buen estado clínico en el momento del diagnóstico, a menudo se opta por una fase inicial de observación [1,2]. Así, muchos pacientes pueden llevar una vida normal con relativamente pocos síntomas durante algunos años. Con esta estrategia, sólo se interviene si se ha producido una transformación en una neoplasia maligna de grado superior (grado III o IV) o si la neoplasia maligna ha alcanzado un grado superior (grado III o IV). si se manifiestan síntomas neurológicos hasta signos de presión cerebral (cefalea, trastornos de la vigilancia, náuseas hasta vómitos). Entonces se programa la reducción de la masa, seguida de radioquimioterapia, o se renuncia.
Craneotomía despierto y neuromonitorización
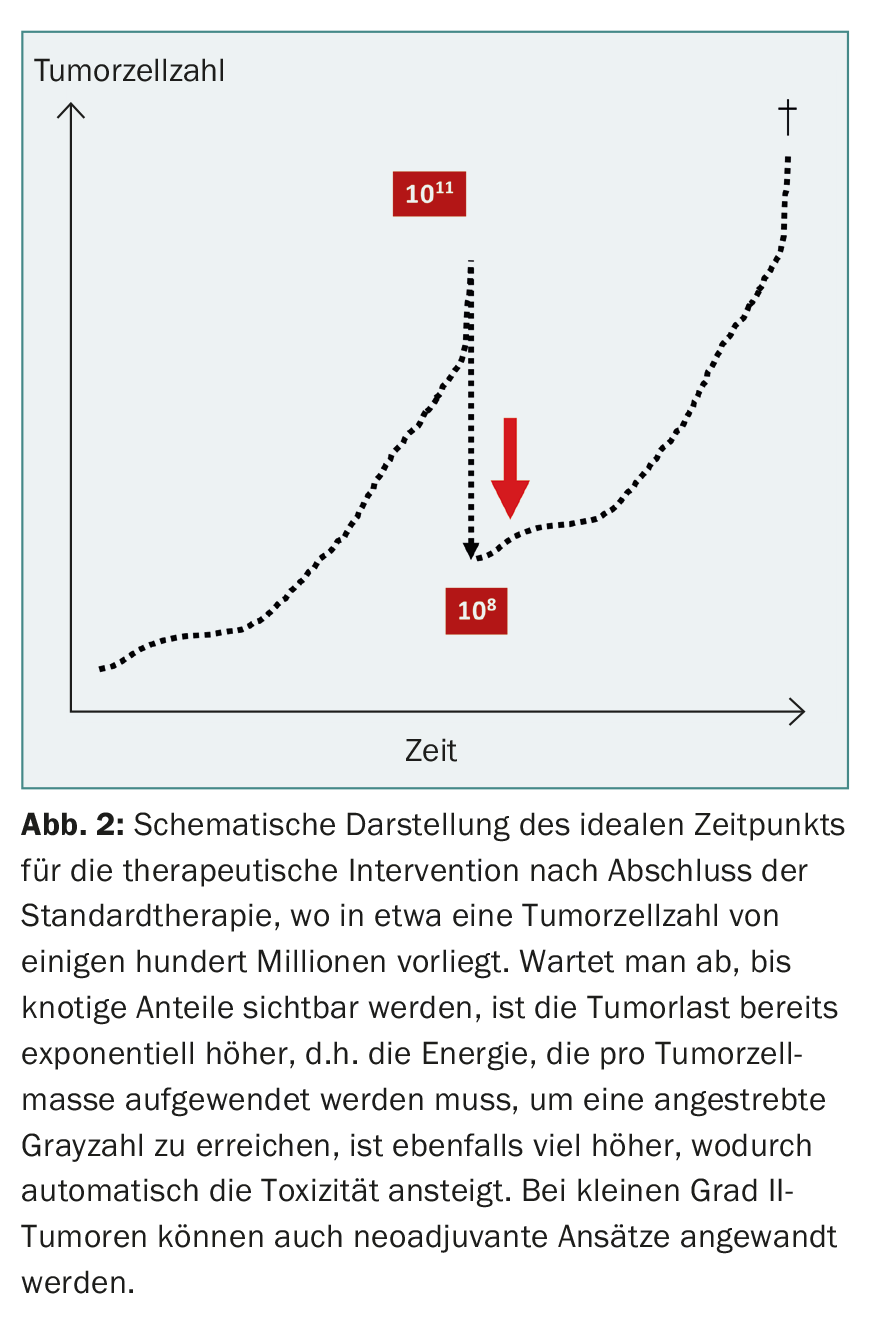
En la fase inicial de la cirugía del glioma, también se intentó en casos desesperados controlar el tumor mediante hemisferectomía, lo que no tuvo éxito debido a la tendencia a la infiltración difusa de las células tumorales. En los últimos años, la cirugía agresiva del glioma ha experimentado un renacimiento, aunque limitado a la reducción extensiva de la masa mediante craneotomía despierta y neuromonitorización [8]. La reorganización cortical debida a la plasticidad cerebral permite la resección ocasional de zonas funcionalmente importantes sin déficits adicionales. Lógicamente, este enfoque tampoco puede conducir a la curación, ya que millones de células tumorales infiltradas permanecen sin detectar. Esto puede demostrarse con un modelo de cálculo: Un cuerpo humano con un peso corporal de 70 kg consta de aproximadamente1014 células. Por tanto, un tumor de 70 g contiene aproximadamente 1011 células tumorales. Con una resección del 99,9 % de una neoplasia infiltrante, aún quedan unos 100 millones de células tumorales, que determinan el destino posterior (Fig. 2) . Una terapia moderna tiene que eliminar estas células residuales o impedir que se desarrollen. puede controlar.
Perspectivas terapéuticas
Muchos pacientes con astrocitoma de grado II no superan los 50 años o no viven más allá de esa edad. sólo con déficits neurológicos crecientes y están expuestos a la amenaza constante del 50% de probabilidad de transformación a un nivel de malignidad superior. Esto habla en favor de la intervención temprana. La resección supramáxima no conduce a la curación, pero reduce el riesgo de transformación maligna. La experiencia inicial con la irradiación unicelular dirigida utilizando biomoléculas difusibles que se acoplan a receptores específicos de células tumorales y portan un isótopo de muy corto alcance y alta energía como efector ha conducido repetidamente a un control tumoral muy prolongado, muy por encima de la supervivencia media, sin toxicidad significativa [9,10]. En el futuro se añadirán otros fármacos que bloqueen los circuitos reguladores biológicos alterados y mejoren así el pronóstico [5].
Mensajes para llevarse a casa
- Alrededor del 10-15% de todos los gliomas malignos se presentan principalmente como astrocitomas de grado II poco malignos o, con menor frecuencia, como oligodendrogliomas. La supervivencia media es de siete a diez años para los astrocitomas de grado II y de diez a 15 años para los oligodendrogliomas de grado II.
- Teniendo en cuenta el pésimo pronóstico de los glioblastomas más comunes, los gliomas de grado II tienden a subestimar su malignidad en la fase inicial.
- Los tumores cerebrales son probablemente una consecuencia de la tasa de mutación espontánea de los procesos celulares, en cierto sentido la otra cara de la recombinación.
- La nueva clasificación de la OMS de los tumores cerebrales basada en el patrón de metilación y el alelotipo permite una afirmación fiable sobre el pronóstico.
- No existe ninguna norma para el tratamiento de los gliomas de grado II. Además del enfoque de esperar y ver, cada vez se adoptan más tácticas quirúrgicas agresivas. Sin embargo, debido a la infiltración de células tumorales en el tejido cerebral sano, no se puede conseguir la curación. Existen los primeros indicios de que la radioterapia unicelular dirigida con biomoléculas difusibles podría mejorar significativamente el pronóstico.
Literatura:
- Merlo A, de Tribolet N: Tumores cerebrales y de la médula espinal. En: Steck A, Hess CH (eds.): Fisiopatología neurológica. Berna: Verlag Hans Huber 2003.
- Schneider T, et al: Gliomas en adultos. Dtsch Arztebl Int 2010; 107(45): 799-807.
- Merlo A, Rochlitz C, Scott R: Supervivencia de pacientes con síndrome de Turcot y glioblastoma [letter]. N Engl J Med 1996; 334(11): 736-737.
- Merlo A, Bettler B: Glioblastomas en movimiento. Ciencia STKE 2004; 2004(229): pe18.
- Lino M, Merlo A: Trasladar la biología a la clínica: el caso del glioblastoma. Curr Opin Cell Biol 2009; 21(2): 311-316.
- Louis DN, et al: Clasificación de los tumores del sistema nervioso central de la Organización Mundial de la Salud de 2016: resumen. Acta Neuropathol 2016; 131(6): 803-820.
- Linz U: Comentario sobre los efectos de la radioterapia con temozolomida concomitante y adyuvante frente a la radioterapia sola sobre la supervivencia en el glioblastoma en un estudio aleatorizado de fase III: análisis a 5 años del ensayo EORTC-NCIC (Lancet Oncol. 2009;10:459-466). Cáncer 2010; 116(8): 1844-1846.
- Duffau H: La justificación de realizar una resección precoz en el glioma difuso incidental de bajo grado: hacia una “neurooncología quirúrgica preventiva”. Neurocirugía Mundial 2013; 80(5): e115-e117.
- Cordier D, et al: Terapia dirigida con radionucleidos alfa de gliomas localizados funcionalmente de forma crítica con 213Bi-DOTAGA-sustancia P: un ensayo piloto. Eur J Nucl Med Mol Imaging 2010; 37(7): 1335-1344.
- Cordier D, et al: Compuestos radiomarcados dirigidos en la terapia del glioma. Semin Nucl Med 2016; 46(3): 243-249.
InFo ONCOLOGÍA Y HEMATOLOGÍA 2017; 5(6): 7-10