Si se altera la formación de nuevas células sanguíneas en la médula ósea, la causa puede ser una forma rara de cáncer crónico de la sangre. La hiperproliferación de las tres series celulares de la médula ósea provoca eritrocitosis, trombocitosis y leucocitosis en la policitemia vera. El resultado es, entre otras cosas, un aumento significativo de los niveles de hematocrito y, por tanto, también del riesgo de episodios tromboembólicos.
La policitemia vera (PV) es una neoplasia mieloproliferativa crónica muy poco frecuente y se caracteriza por un aumento de la hematopoyesis. La mayoría de los pacientes con PV presentan una mutación en el gen de la tirosina quinasa JAK2 [1]. Da lugar a un aumento de la proliferación celular, así como de la producción de citoquinas proinflamatorias. La sobreproducción de eritrocitos y el consiguiente aumento del hematocrito incrementan la viscosidad de la sangre. De este modo, se favorece la aparición de tromboembolias: el 45% de todas las muertes en PV se deben a complicaciones tromboembólicas [2]. Sin embargo, el pronóstico suele ser favorable. La edad media en el momento del diagnóstico es de 65 años.
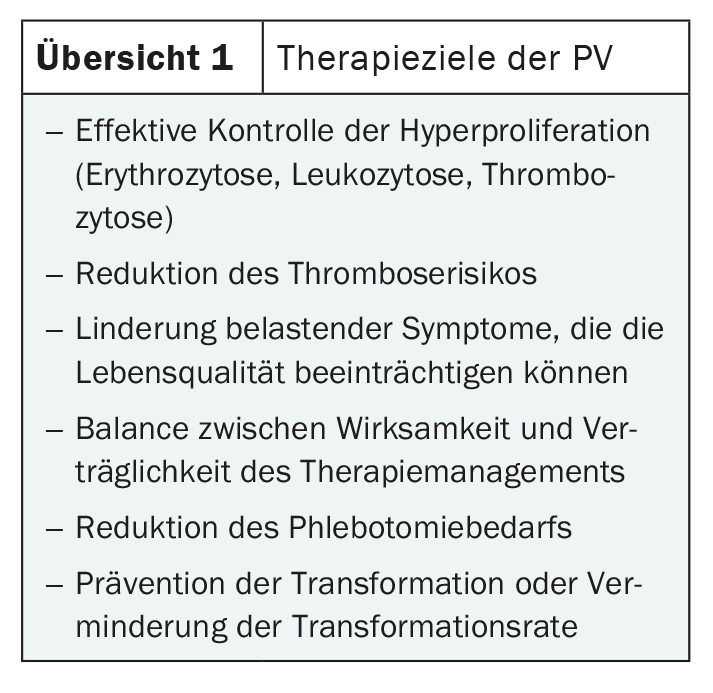
Aunque los síntomas son variados, suelen ser bastante inespecíficos. Por lo tanto, el diagnóstico a menudo se hace sólo por casualidad. Los posibles síntomas incluyen dolores de cabeza, alteraciones visuales, fatiga y prurito, así como dolor óseo y dolor en la parte superior del abdomen. A menudo están causadas por la esplenomegalia típica de la PV [1,2]. El aumento de la masa celular sanguínea puede provocar trastornos circulatorios que pueden conducir a trombosis venosas y arteriales graves como embolia pulmonar, apoplejía o infarto de miocardio. Por lo tanto, está indicado un diagnóstico precoz y un tratamiento eficaz (Visión general 1) [3].
En la fase crónica, que suele durar años, las características clínicas de aumento de la mieloproliferación cobran protagonismo. Las complicaciones más frecuentes y potencialmente amenazadoras son el tromboembolismo arterial o venoso en hasta un 40% de los pacientes. En la fase tardía de la enfermedad, el principal problema radica en la llamada fase “retardada”. Se caracteriza por una disminución de la eritrocitosis y un aumento de la esplenomegalia, combinado con una fibrosis de la médula ósea, que puede ir seguida de una transformación en mielofibrosis post-PV (secundaria) y/o leucemia aguda [1]. La tasa global de FPM post-PV es de alrededor del 15% tras un periodo medio de observación de 10 años y del 50% tras 20 años.
Tratamiento adaptado al riesgo
Dado que la prevención de la tromboembolia es primordial, la flebotomía suele considerarse el tratamiento de elección. Esto puede bajar el hematocrito (Hct) por debajo del 45% y reducir la hiperviscosidad de la sangre. Los estudios han demostrado que fijar la Hct por debajo del 45% puede reducir la tasa de muerte cardiovascular en PV a una cuarta parte [4]. Sin embargo, una flebotomía es muy agotadora. Por lo tanto, también debe iniciarse inicialmente un tratamiento con dosis bajas de ácido acetilsalicílico (AAS). La recomendación de tratamiento se basa entonces en la puntuación de riesgo. Puede suponerse un riesgo bajo para los pacientes más jóvenes <60 años que no hayan sufrido una trombosis anteriormente. Actualmente se está debatiendo si el tratamiento reductor de citocinas también debería considerarse para ellos en determinadas condiciones.

Sin embargo, la mayoría de los pacientes con PV corren un alto riesgo de todos modos. En estos casos, está indicado iniciar una terapia citorreductora. La hidroxiurea (HU) o el interferón alfa (INF) se recomiendan para el tratamiento primario [5]. Sin embargo, la HU en particular no es adecuada para todos los pacientes y puede causar efectos secundarios graves (Resumen 2) [6]. Por lo tanto, en el caso de las pacientes más jóvenes que desean tener hijos, es más probable que se utilice la INF. Si la terapia de primera línea no se tolera o los síntomas clínicos no remiten lo suficiente, debe cambiarse el tratamiento. El inhibidor de JAK2 ruxolitinib ha demostrado en estudios controlar el aumento de la mieloproliferación a la vez que es bien tolerado [1]. También desaparecieron muchos síntomas asociados a la PV, como la fatiga y el prurito. Además, la mayoría de los pacientes experimentaron el efecto muy rápidamente, en las primeras cuatro semanas. El busulfán puede utilizarse como terapia alternativa en pacientes de edad avanzada. Sin embargo, en este caso siempre se discute el potencial leucemógeno, por lo que la sustancia sólo debe utilizarse con moderación. La anagrelida puede considerarse un compañero de combinación de, por ejemplo, HU o INF. Está destinado exclusivamente a reducir la producción de plaquetas y puede actuar como complemento si no se obtienen resultados satisfactorios con las otras sustancias por sí solas.
Literatura:
- www.onkopedia.com/de/onkopedia/guidelines/polycythaemia-vera-pv/@@guideline/html/index.html (última llamada el 23/04/2024).
- Vannucchi AM, et al.: N Engl J Med 2015; 372: 426–435.
- Stein BL, Moliterno AR, Tiu RV: Polycythemia vera disease burden: contributing factors, impact on quality of life, andemerging treatment options. Ann Hematol 2014; 93: 1965–1976.
- Marchioli R, Finazzi G, Specchia G, et al.: Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013; 368: 22–33.
- Barbui T, Tefferi A, Vannucchi AM, et al.: Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European Leukemia Net. Leukemia 2018; 32: 1057–1069.
- Barosi G, Birgegard G, Finazzi G, et al.: A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol 2010; 148: 961–963.
InFo ONKOLOGIE & HÄMATOLOGIE 2024; 12(2): 38









