El Grupo de Trabajo sobre enfermedades ampollosas autoinmunes de la Academia Europea de Dermatología y Venereología (EADV) ha publicado recientemente nuevas recomendaciones para el tratamiento del penfigoide ampolloso. La evaluación consensuada de los expertos se basa en las pruebas clínicas actuales relativas a los métodos de diagnóstico y las estrategias terapéuticas.
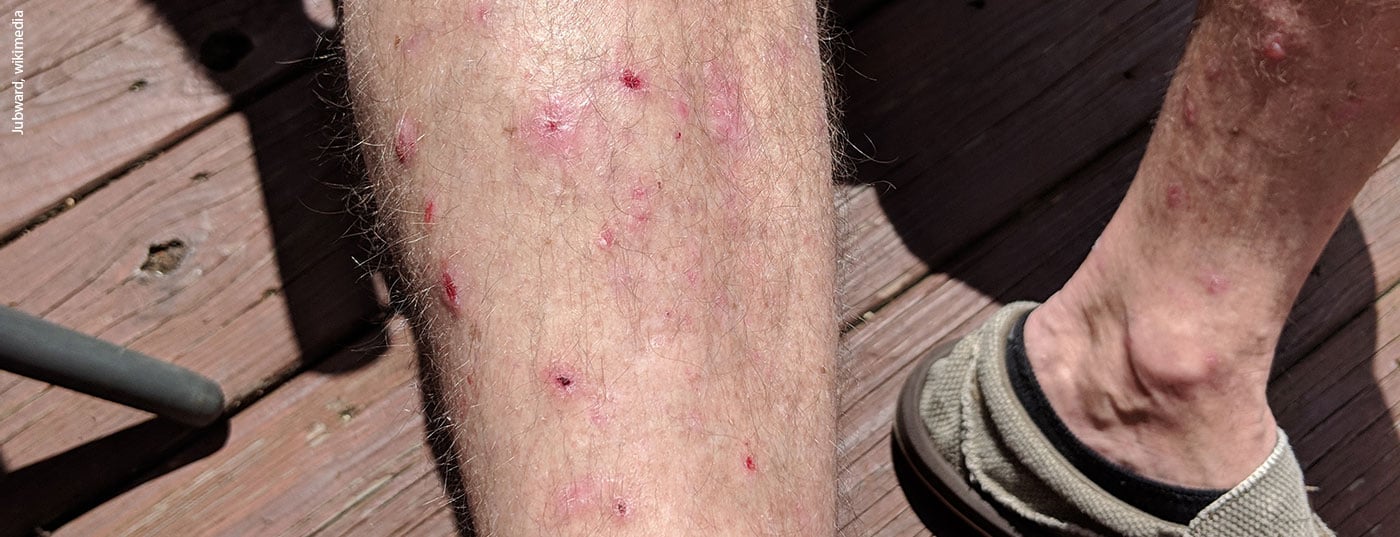
Las enfermedades autoinmunes ampollosas son enfermedades raras pero potencialmente mortales de la piel y las mucosas. El penfigoide bulloso es la dermatosis autoinmune bullosa más común. Se forman autoanticuerpos contra los hemidesmosomas de los queratinocitos basales con formación de ampollas subepidérmicas [1]. La enfermedad suele aparecer en pacientes de edad avanzada. Son características las lesiones bullosas localizadas o generalizadas sobre piel inflamada enrojecida o normal [2]. Un subgrupo de pacientes desarrolla únicamente excoriaciones, lesiones pruriginosas y lesiones eritematosas eccematosas y/o urticariales. Casi todos los pacientes sufren picores pronunciados [3]. El penfigoide bulloso es una enfermedad que se asocia a una elevada morbilidad y afecta significativamente a la calidad de vida.
En la directriz actualizada, los pasos diagnósticos se describen detalladamente con referencia a los últimos hallazgos [2]. Hoy en día se dispone de un gran arsenal de tecnologías de vanguardia.

Microscopía de inmunofluorescencia indirecta y ELISA
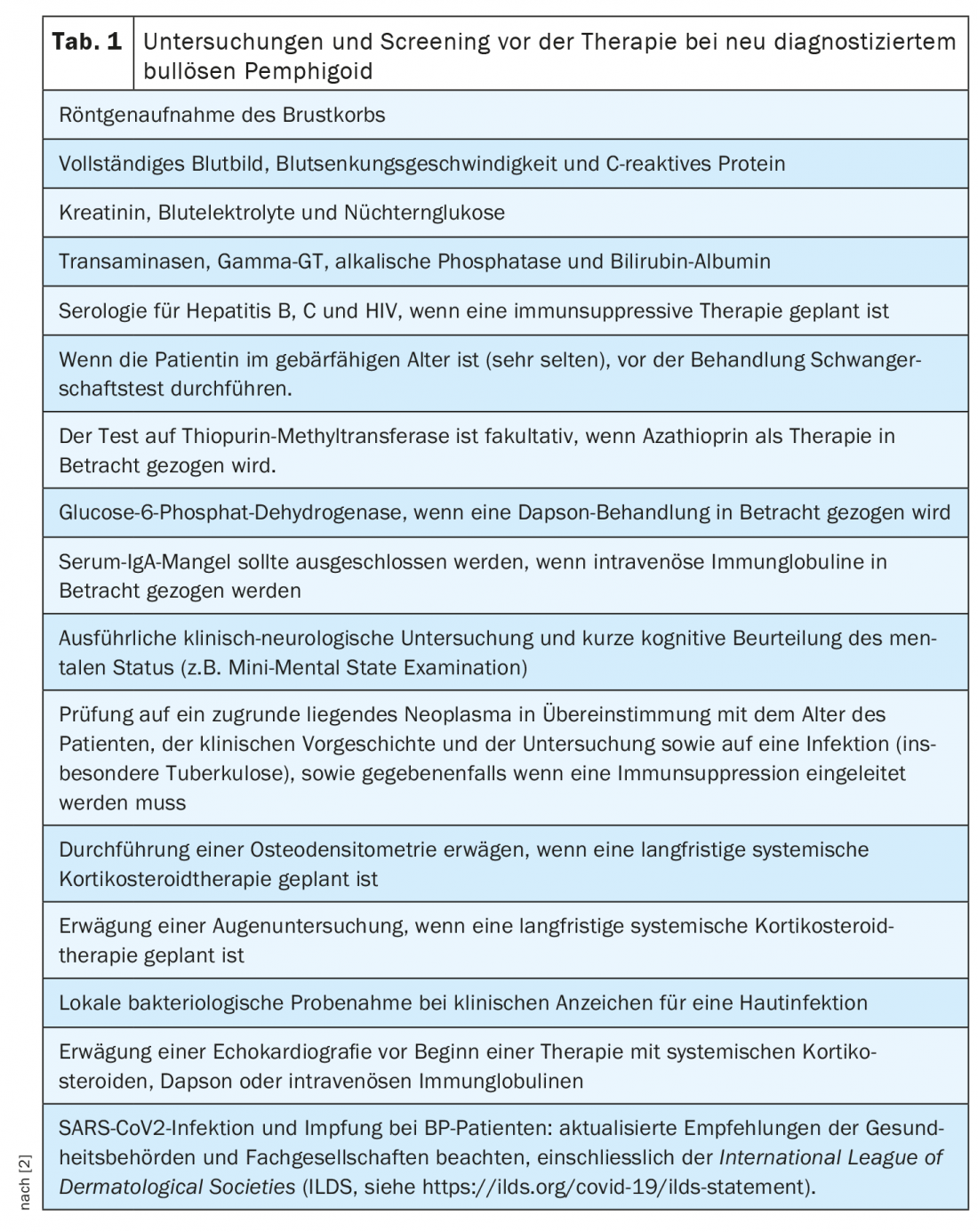
Además de la anamnesis y la exploración física -incluido el registro del BPDAI (“Bullous Pemphigoid Disease Area Index”), la calidad de vida y cualquier comorbilidad presente- existe una amplia gama de pruebas de laboratorio y otros procedimientos de pruebas inmunopatológicas que pueden utilizarse (resumen 1) [2]. El diagnóstico del penfigoide bulloso se basa en una combinación de características clínicas y hallazgos positivos en la microscopía de inmunofluorescencia directa e indirecta. Los autoanticuerpos circulantes pueden detectarse mediante ELISA y microscopía de inmunofluorescencia indirecta. El análisis de los depósitos lineales a lo largo del límite dermoepidérmico es un enfoque práctico fiable para diferenciar el penfigoide bulloso de otras formas de penfigoide. A la hora de elegir la opción terapéutica adecuada, también deben tenerse en cuenta enfermedades concomitantes como la hipertensión. Las investigaciones preliminares recomendadas para los pacientes con penfigoide bulloso recién diagnosticado se resumen en la tabla 1 .
Propionato de clobetasol tópico como principal componente del tratamiento
Las recomendaciones de consenso aconsejan el uso de corticosteroides tópicos de alta potencia siempre que sea posible [2]. Para ello se recomienda la crema de propionato de clobetasol al 0,05%, ambas para infestaciones localizadas, limitadas y moderadas, cada una en una dosis de 20-30 g diarios. En casos graves, puede utilizarse una dosis de 30-40 g/día, inicialmente una o dos veces al día sobre todo el tegumento, incluidas las zonas sanas de la piel, excluyendo la cara. Como alternativa, puede utilizarse prednisolona oral. En estudios observacionales prospectivos, la prednisolona a una dosis inicial de 0,5 mg/kg/día el día 21 dio lugar al control de la enfermedad en aproximadamente dos tercios de los pacientes con penfigoide ampolloso de leve a moderado, y a una enfermedad grave en sólo el 46% de los casos [11]. El control de la enfermedad se define como un estado en el que no aparecen nuevas lesiones ni síntomas de picor y las lesiones existentes se curan. En los casos graves, la prednisolona 1 mg/kg/d ha demostrado ser eficaz, pero esta terapia de dosis alta se asocia a un mayor riesgo de efectos secundarios y mortalidad en comparación con la terapia tópica de gran superficie con propionato de clobetasol al 0,05%. Por lo tanto, no se recomienda la prednisolona 1 mg/kg/d como tratamiento inicial [2].
En caso de contraindicaciones o de respuesta insuficiente a los corticosteroides, pueden utilizarse inmunosupresores como el metotrexato, la azatioprina, el mofetil micofenolato o el ácido micofenolato. El uso de doxiciclina y dapsona es controvertido y se limita principalmente al uso adyuvante en combinación con corticosteroides tópicos.
Se recomienda el uso de aditivos antisépticos para el baño como medida complementaria a la terapia farmacológica. Si hay lesiones erosivas extensas, pueden cubrirse con apósitos, preferiblemente no adherentes para reducir la sobreinfección bacteriana y el dolor y favorecer la cicatrización.
Biológicos como complemento o monoterapia
En los casos difíciles de tratar de penfigoide ampolloso, puede considerarse el uso de productos biológicos como opción de tratamiento única o adicional, según las directrices. Los factores a tener en cuenta incluyen las características clínicas, el curso previo, la respuesta al tratamiento y las contraindicaciones al tratamiento estándar. Los productos biológicos se dirigen a las citocinas proinflamatorias y otras dianas celulares que contribuyen al daño tisular en el penfigoide bulloso.

Entre los anticuerpos monoclonales potencialmente eficaces se encuentran el rituximab (anti-CD20), el omalizumab (anti-IgE) y el dupilumab (anti-IL-4/-13). Las recomendaciones de los expertos a este respecto se resumen en el recuadro [2]. También se están investigando otros enfoques terapéuticos, como el bloqueo de IL-17, IL-12/-23, IL-5Ra [4]. Además, existen pruebas empíricas de que el uso de inmunoglobulina intravenosa (2 g/kg/d) como complemento es eficaz en los casos refractarios, con efectos secundarios potencialmente graves en los pacientes de edad avanzada.
Seguimiento del curso de la terapia
El penfigoide bulloso conlleva riesgos de complicaciones en relación con la propia enfermedad, pero también en relación con los efectos secundarios relacionados con la terapia. Para evaluar la eficacia y la seguridad de una terapia, se recomiendan exámenes periódicos de seguimiento [2]. La frecuencia de las citas de seguimiento depende de varios factores. Hasta que se logre el control de la enfermedad, se recomiendan controles a intervalos de 1-2 semanas, luego a intervalos de cuatro semanas durante tres meses, y después cada 2-4 meses al final del tratamiento.
Literatura:
- Joura MI, et al: Dermatología geriátrica. Z Gerontol Geriat 2022, https://doi.org/10.1007/s00391-021-02006-2, (última consulta: 19.08.2022).
- Borradori L, et al: Directrices S2 K actualizadas para el tratamiento del penfigoide ampolloso iniciadas por la Academia Europea de Dermatología y Venereología (EADV). J Eur Acad Dermatol Venereol. 2022 Jun 29. doi: 10.1111/jdv.18220. Publicación electrónica antes de impresión.
- UKSH: Dermatosis autoinmunes, www.uksh.de/dermatologie-luebeck
- Clinicaltrials.gov: https://clinicaltrials.gov/ct2, (última consulta: 19.08.2022)
- Schmidt E, et al: Rituximab en enfermedades ampollosas autoinmunes: respuestas mixtas y efectos adversos. Br JDermatol 2007; 156: 352-356.
- Hall RP 3rd, et al: Association of serum B-cell activating factor level and proportion of memory and transitional B cells with clinical response after rituximab treatment of bullous pemphigoid patients.
- J Invest Dermatol 2013; 133: 2786-2788.
- Joly P: Grupo de estudio francés sobre enfermedades cutáneas ampollosas autoinmunes, la red francesa de enfermedades raras en D. Incidencia y gravedad de COVID-19 en pacientes con enfermedades cutáneas ampollosas autoinmunes: un estudio a escala nacional. J Am Acad Dermatol 2021; 86: 494-497.
- Abdat R, et al: Dupilumab como terapia novedosa para el penfigoide bulloso: una serie de casos multicéntrica. J Am Acad Dermatol 2020; 83: 46-52.
- Seyed Jafari SM, et al: Informe de un caso: combinación de omalizumab y dupilumab para el penfigoide bulloso recalcitrante. Front Immunol 2020; 11: 611549.
- Fairley JA, et al: Patogenicidad de la IgE en la autoinmunidad: éxito del tratamiento del penfigoide bulloso con omalizumab. J Allergy Clin Immunol 2009; 123: 704-705.
PRÁCTICA DERMATOLÓGICA 2022; 32(4): 31-32









