Una eosinofilia sanguínea se define por un número >500 eosinófilos por μl. Si se mide una cifra >1500/μl al menos dos veces en dos semanas, se está ante una hipereosinofilia (HE). El síndrome hipereosinofílico (SHE) se produce cuando a la HE se añaden daños orgánicos. En el Congreso DGIM 2023 se debatió, por ejemplo, cómo clasificar mejor la HES y diagnosticar la EGPA.
Desde 2012, la ES y la HES se subdividen en función de las diferentes etiologías en formas primarias y formas reactivas, así como en ES y HES idiopáticas en las que no se detecta ni una enfermedad subyacente clonal ni reactiva. Debido al creciente número de marcadores y objetivos, a principios de este año se publicó una actualización de esta clasificación [1]. Entre otras cosas, la actualización estipula que se requiere un recuento medido de >1500/µl en al menos dos ocasiones con un intervalo de al menos dos semanas para definir la ES – en la versión anterior, el intervalo de tiempo seguía siendo de cuatro semanas.
Como escriben el Prof. Dr. Peter Valent, del Hospital Universitario AKH de Viena, y sus colegas, además de la presencia de HE en sangre y/o tejidos y el daño orgánico asociado a la HE como tercer criterio, la HES se define por la exclusión de otra enfermedad o patología subyacente como causa primaria del daño orgánico.
Los autores subrayan que el HES no es un diagnóstico definitivo ni una enfermedad inmunológica o hematológica definida. Más bien, debe identificarse la etiología contribuyente en todos los pacientes con HES, si es posible. Si no se identifica ninguna enfermedad subyacente, el diagnóstico final es HES idiopática (HESI).
Los subtipos de HES son trastornos mieloproliferativos (14%) y linfoproliferativos (14%), idiopáticos (42%), asociados (9%) y superpuestos (21%). El grupo de enfermedades asociadas también incluye la granulomatosis eosinofílica con poliangitis (EGPA), antes conocida como síndrome de Churg-Strauss.
Los anticuerpos citoplasmáticos antineutrófilos (ANCA) se detectan en aproximadamente el 40% de los pacientes con EGPA. Dada la similitud de las manifestaciones clínicas de la EGPA (y otros síndromes relacionados con la HE) con las características clásicas de la HES, Valent et al. concluyó que estos síndromes deben clasificarse como HES si se cumplen los criterios de HES. Si no es así, el diagnóstico final debe ser el síndrome correspondiente y no el HES.
| Criterios de clasificación ACR para vasculitis Eosinofilia >10% (opcional) Eosinofilia tisular Asma (>90%) Anomalías NNH (aprox. 50%) Infiltrados pulmonares volátiles (50-70%) Neuropatía (aprox. 75%) |
| según [3] |
A menudo no es posible detectar la vasculitis
“Para distinguir entre una HES y una EGPA, hay que saber que la EGPA necesita pruebas de vasculitis”, explicó el Dr. Christof Iking-Konert, jefe del departamento de reumatología del Stadtspital Triemli de Zúrich [2]. Se refirió a esto. Según los criterios de clasificación del Colegio Americano de Reumatología (ACR) [3] (recuadro).
La EGPA es la forma más rara de vasculitis asociada a ANCA, con una prevalencia de 24/1 millón, por lo que el primer reto diagnóstico es pensar en ella antes. El segundo obstáculo: La evidencia requerida de vasculitis en la histología es a menudo difícil porque, por ejemplo, una biopsia no es factible en pacientes muy enfermos. Por lo general, la EGPA sólo se sospecha, por lo que se utilizan sustitutos de la vasculitis, como explicó la Dra. Iking-Konert. Según él, si uno o más de estos factores están presentes, se puede suponer un correlato de vasculitis:
- Hemorragia alveolar
- Púrpura palpable
- Infarto de miocardio debido a coronariitis probada
- Hematuria asociada a recuentos de glóbulos rojos o >10% de glóbulos rojos dismórficos o hematuria y proteinuria 2+.
- Mononeuritis o mononeuritis múltiple
- MPO ANCA y cualquier tipo de manifestación sistémica.
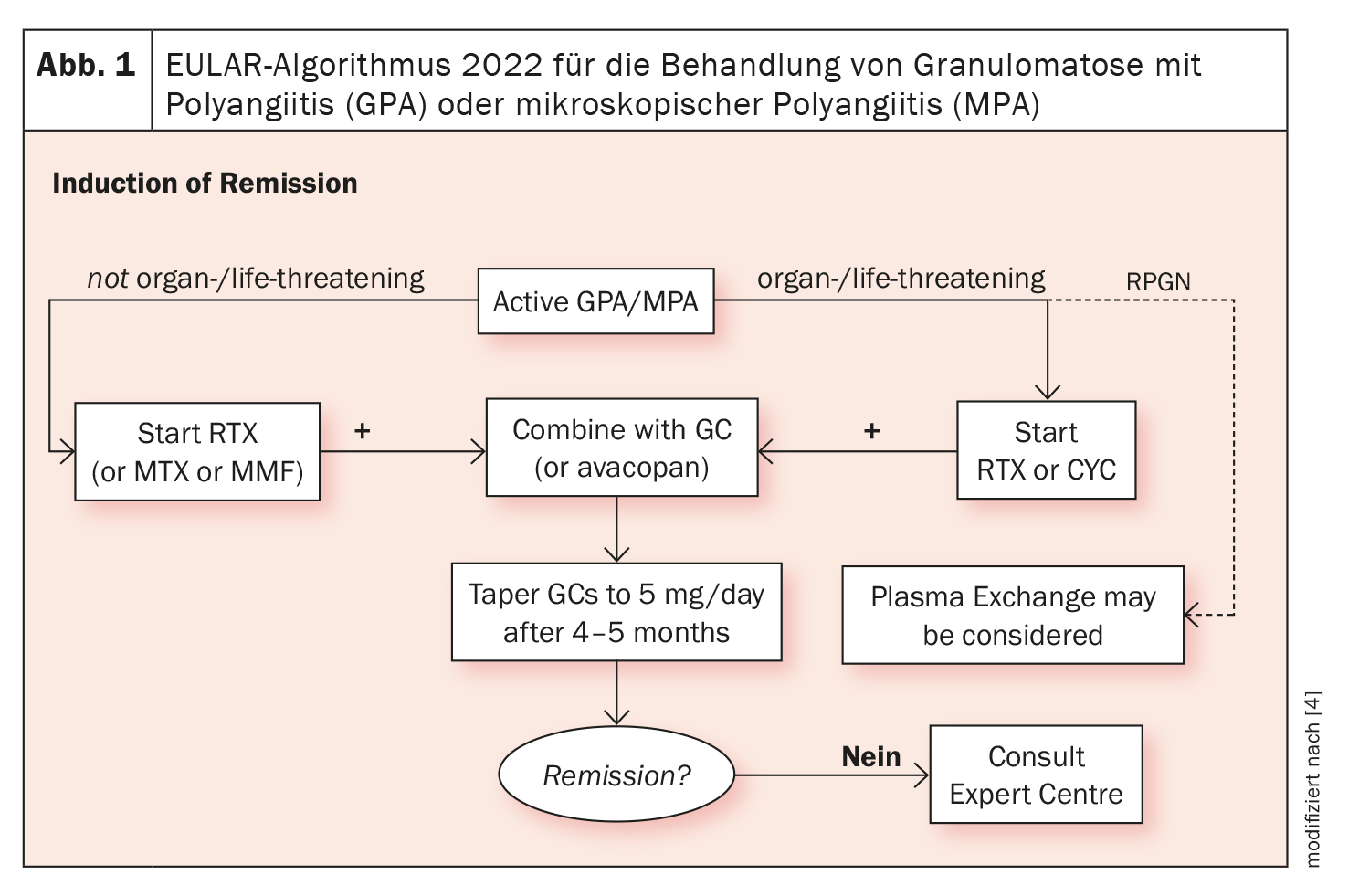
Los productos biológicos están disponibles para el tratamiento de la EGPA desde hace más de una década. Además del rituximab, se han establecido terapias dirigidas con anticuerpos contra la IL-5. La Dra. Iking-Konert se refirió a la directriz EULAR sobre el tratamiento de la vasculitis asociada a ANCA, que se actualizará en 2022: En ella se recomienda iniciar una terapia con ciclofosfamida (CYC) o rituximab (RTX), que se combinan con cortisona, en caso de EGPA con peligro para los órganos o la vida. En las enfermedades no orgánicas o potencialmente mortales, el bloqueo de la IL-5 está indicado para los cuadros refractarios o graves (Fig. 1 ) [4].
Congreso: DGIM Industriesymposium GSK
Literatura:
- Valent P, et al.: Allergy 2023; 78: 47–59.
- Industrie Symposium «Und täglich grüsst das Immunsystem: seltene Autoimmunerkrankungen erkennen und behandeln». 129. Kongress der DGIM, 23.04.2023; Veranstalter: GSK.
- Jennette JC, et al.: Arthritis Rheum 1994; 37: 187–192.
- Hellmich B, et al.: Ann Rheum Dis 2023; doi: 10.1136/ard-2022-223764.
| Imagen de portada: Micrografía a gran aumento de una vasculitis eosinofílica compatible con el síndrome de Churg-Strauss, abreviado CSS. H&E stain. Quelle: Nephron, wikimedia |
InFo RHEUMATOLOGIE 2023; 5(1): 17