Las neoplasias neuroendocrinas suelen aparecer de forma esporádica y a menudo se originan en el espacio gastroenteropancreático. Suelen diagnosticarse de forma incidental, ya que a menudo no son funcionales. Por lo tanto, en la mayoría de los casos se trata de una enfermedad tumoral ya avanzada, a menudo metastásica.
El término “neuroendocrino” hace referencia a células con características histológicas de tejido glandular que suelen estar presentes de forma difusa en diversos órganos del cuerpo y pueden segregar hormonas. Según la función y la localización, se producen aminas (por ejemplo, catecolaminas) o péptidos (por ejemplo, somatostatina) que regulan la actividad del tejido circundante. Embriológicamente, durante mucho tiempo se consideraron parte de la cresta neural, pero ahora se clasifican como parte del endodermo, al igual que las células exocrinas del tracto gastrointestinal. Históricamente, estas células se agrupaban en un sistema neuroendocrino difuso (antes también llamado APUD (“captación y descarboxilación de precursores de aminas”)). Sin embargo, dado que estos términos carecen de significado histopatológico o clínico relevante, deberían dejar de utilizarse.
Si estas células degeneran, se denominan neoplasias neuroendocrinas (NEN). Este término general incluye tanto los tumores neuroendocrinos (NET) de proliferación lenta como los carcinoides de pulmón, así como los carcinomas neuroendocrinos (NEC), mucho más agresivos y de pronóstico desfavorable. Así pues, todas las recomendaciones de este artículo se basarán en esta clasificación.
Las neoplasias neuroendocrinas se producen sobre todo de forma esporádica, con una incidencia estimada de aproximadamente 5/100.000 habitantes. Puede observarse un cluster familiar en la neoplasia endocrina múltiple (MEN) I y II, así como en enfermedades raras como el síndrome de von Hippel-Lindau o la neurofibromatosis tipo 1 (Recklinghausen). La mayoría de los NEN se originan en el espacio gastroenteropancreático (GEP-NEN), alrededor del 25-30% se originan en el pulmón [1].
Sintomatología
Aunque la producción de hormonas es una característica típica de las neoplasias neuroendocrinas, los llamados tumores funcionales -es decir, tumores cuya producción y secreción de hormonas provoca síntomas- son poco frecuentes. Las molestias causadas por los tumores funcionales dependen de la secreción respectiva de sustancias bioactivas. Los insulinomas o gastrinomas, que representan la mayor parte de estos NEN funcionales, provocan, por ejemplo, hipoglucemias graves, potencialmente mortales, o úlceras gástricas múltiples (síndrome de Zollinger-Ellison). Otro cuadro clínico típico es el síndrome carcinoide, en el que la secreción excesiva de serotonina puede provocar diarrea acuosa grave, síntomas de rubor y broncoespasmo. La fibrosis endocárdica puede producirse como complicación tardía, lo que puede llevar a una insuficiencia cardiaca con consecuencias mortales (síndrome de Hedinger).
Sin embargo, hasta el 90% de todos los NEN son no funcionales y se diagnostican como hallazgos incidentales o en el contexto de dolencias inespecíficas. En la mayoría de los casos, debido a su curso indolente, se trata de una enfermedad tumoral avanzada, ya metastatizada.
Diagnóstico
Siempre es necesaria una biopsia para confirmar el diagnóstico. El estudio histopatológico debe incluir la determinación de los marcadores típicos (sinaptofisina, receptores de somatostatina [SSTR]) y necesariamente el índice de proliferación (Ki-67 o MIB-1) o el índice de mitosis, ya que tienen una importancia considerable tanto pronóstica como terapéutica. En caso de sospecha de síndrome carcinoide, la determinación de ácido 5-hidroxiindolacético (5-HIESS, un producto de degradación de la serotonina) en la orina de 24 horas es diagnóstica. Otros valores de laboratorio en ayunas, como la cromogranina A (CgA) y, en caso necesario, la enolasa neuronal específica (NSE), pueden utilizarse como marcadores tumorales. Dependiendo de la clínica, se utilizan otras pruebas (por ejemplo, la prueba de ayuno para el insulinoma). Como perspectiva de futuro, también hay que mencionar aquí un nuevo método: el llamado NETest [2]. Se trata de un diagnóstico por PCR a partir de sangre total (“biopsia líquida”), con el que se examinan 51 cadenas de ARNm específicas de NET. El valor predictivo positivo para el diagnóstico de neoplasia neuroendocrina es superior al 90%, al igual que la sensibilidad y la especificidad. Los informes iniciales han demostrado su utilidad tanto para el diagnóstico como para el seguimiento de la respuesta al tratamiento. En la actualidad, sin embargo, este NETest aún no está ampliamente disponible.
Imágenes
Además de las imágenes habituales, las imágenes nucleares funcionales desempeñan un papel crucial, tanto en la estadificación como en lo que respecta a la selección de la terapia. En primer lugar, cabe mencionar la PET/TC con 68Ga-DOTATATO, que en los últimos años ha sustituido de forma continuada a la centellografía con octreotida, menos precisa y más compleja [3]. Gracias a este examen PET, significativamente más sensible, no es infrecuente que se produzca un aumento de la estadificación mediante la detección de metástasis que no son visibles en la tomografía computarizada. Además, puede utilizarse para detectar la expresión de SSTR2, presente con frecuencia, en la superficie celular de la NET y evaluar así el posible uso de una terapia con radionucleidos (PRRT, véase más adelante). Si el índice de proliferación es elevado o si la expresión de SSTR2 está ausente o es heterogénea, resulta útil la realización adicional de FDG-PET/CT [4], ya que las lesiones ávidas de FDG indican un comportamiento más agresivo y deben examinarse adicionalmente de forma bioptica siempre que sea posible.
Nueva clasificación de la OMS
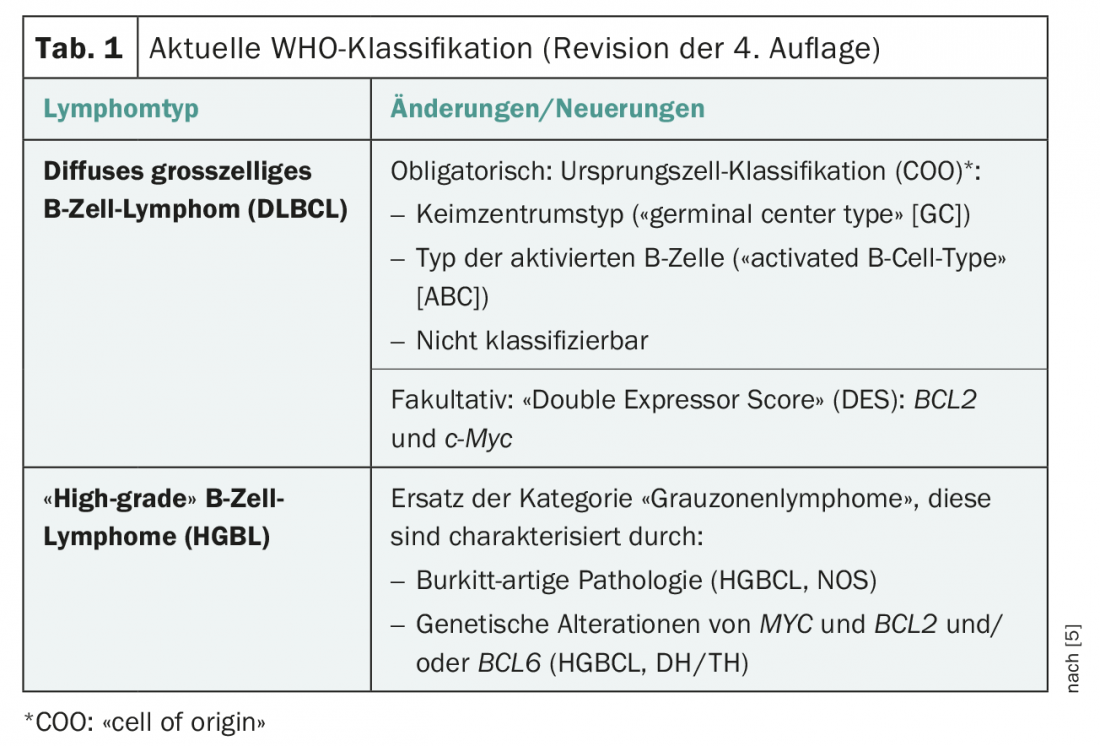
En las últimas décadas se han producido cambios significativos en la clasificación de las neoplasias neuroendocrinas gracias a nuevos hallazgos histopatológicos y clínicos (Tab. 1) . La clasificación actual de la OMS (2017) ha eliminado, en particular para los NEN pancreáticos (nuevo: panNEN), las discrepancias histopatológicas (alto índice de proliferación
pero baja tasa de mitosis) y las características clínicas (pronóstico) se tuvieron en cuenta. El resultado es la subdivisión en NET G1, NET G2, NET G3 y NEC G3. Desde la clasificación de 2010, el término “carcinoide” sólo debe utilizarse para la NET pulmonar (Tab. 2).

Tratamiento de la NET no metastásica y seguimiento
La única opción potencialmente curativa para las neoplasias neuroendocrinas es la extirpación completa del tumor. En el tracto gastrointestinal, puede realizarse por vía endoscópica en función del tamaño (hasta 2 cm) y del situs. Sin embargo, la indicación de resección quirúrgica debe hacerse de bajo umbral si hay evidencia de infiltración de la muscularis propria (T2) o metástasis en los ganglios linfáticos [5]. Las redes del apéndice constituyen una situación especial. En la gran mayoría de los casos, se trata de un hallazgo incidental tras una apendicectomía. La decisión de realizar una hemicolectomía derecha suplementaria con linfadenectomía depende del tamaño del tumor (>2 cm), la profundidad de penetración (>3 mm) en el mesoapéndice y la invasión vascular (V1) o linfática (L1) [6].
A diferencia de otras entidades tumorales, no hay indicación de terapia adyuvante en los tumores neuroendocrinos. Se recomienda un seguimiento regular con endoscopias anuales de NET de estómago o recto. En el caso de una NET localmente avanzada pero extirpada quirúrgicamente por completo, deben realizarse pruebas de imagen anuales (normalmente TC torácica abdominal con medio de contraste i.v.) durante toda la vida.
Opción terapéutica para el VEN metastásico
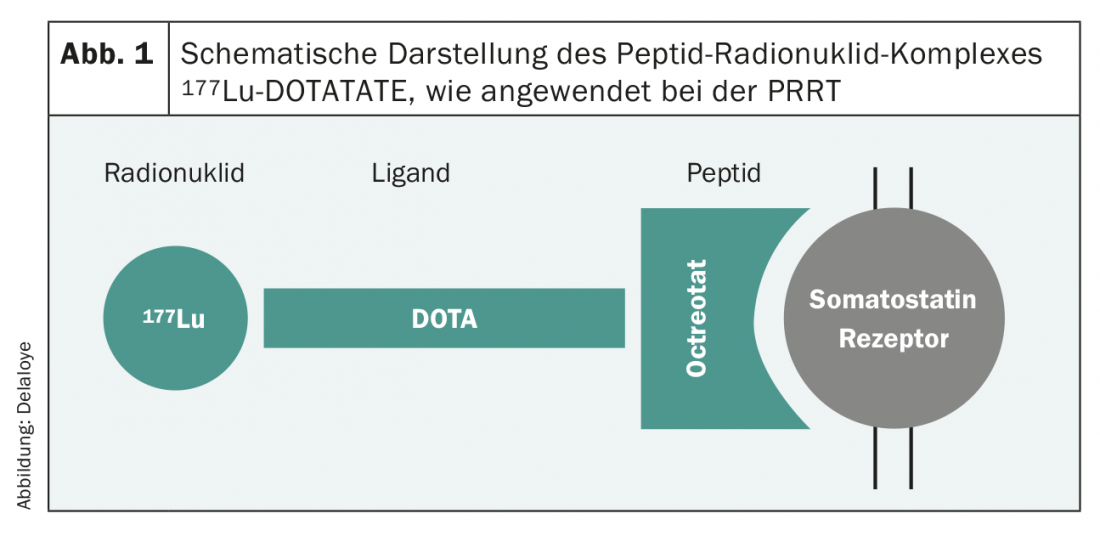
Los nuevos avances de la última década han mejorado drásticamente el panorama terapéutico y, por tanto, la supervivencia de los pacientes. El número de estudios de fase III relevantes para la práctica se ha desarrollado de forma casi exponencial, y las recomendaciones de las sociedades profesionales se basan en pruebas cada vez más sólidas. Básicamente, las opciones de tratamiento para los tumores G1 y G2 son muy similares. En esta población de pacientes, debe prestarse especial atención a los efectos secundarios de la terapia, ya que en su mayoría son pacientes asintomáticos. En los tumores que expresan SSTR2, numerosos estudios han establecido el uso de primera línea de los análogos de la somatostatina [7,8], incluso en el tumor no funcional. Esta terapia, que suele ser bien tolerada, conduce a menudo a un buen control del tumor y, por tanto, a una prolongación de la supervivencia libre de progresión. Otra opción es la terapia con radionucleidos receptores de péptidos (PRRT). Procedimiento en el que una sustancia radiactiva (90Yttrio o 177Lutecio), unida a un péptido afín al receptor de somatostatina (Octreotida/Octreotato) (Fig. 1 ), se administra por vía intravenosa en forma de infusión y permite la irradiación dirigida de las células tumorales SSTR2-positivas. Este tratamiento suele aplicarse cuatro veces seguidas, cada vez con un intervalo de ocho semanas. También se trata de una terapia que suele tolerarse muy bien y cuyos resultados se demostraron hace unos años en un estudio de fase III a gran escala [9].
A nivel molecular, se ha establecido el uso del everolimus como inhibidor de mTOR. El mecanismo de proliferación celular en torno a la vía PI3K-mTOR desempeña un papel importante en las neoplasias neuroendocrinas, al igual que en otros tumores. Así pues, se puede conseguir un buen control tumoral con everolimus [10], pero esta terapia se asocia a un número significativamente mayor de efectos secundarios.
La quimioterapia también se utiliza para los G2-NET y G3-NET más proliferantes. Un estudio de fase II demostró la actividad antitumoral de la capecitabina y la temozolomida (CapTem) en esta cohorte de pacientes [11]. Aunque aún faltan datos de fase III, esta terapia ha demostrado su eficacia en la práctica y se ha establecido rápidamente como alternativa a la anterior combinación de estreptozotocina y 5-fluorouracilo (STZ+5FU). El perfil de efectos secundarios también es favorable en este caso y el manejo es sencillo.
Las neoplasias neuroendocrinas de grado superior (concretamente la NEC G3) se tratan con una combinación de platino y etopósido análoga a la terapia del carcinoma bronquial de células pequeñas. Las nuevas terapias con inhibidores de los puntos de control han tenido poco éxito hasta el momento y, en general, no pueden recomendarse. Se seguirán investigando en ensayos clínicos.
Mensajes para llevarse a casa
- Las células con características histológicas de tejido glandular, que suelen estar presentes de forma difusa en diversos órganos del cuerpo y pueden segregar hormonas, se denominan “neuroendocrinas”. Si degeneran, se denominan neoplasias neuroendocrinas (NEN).
- La mayoría de los NEN se originan en el espacio gastroenteropancreático, alrededor del 25-30% se originan en los pulmones.
- Los tumores funcionales, como los insulinomas o los gastrinomas, son poco frecuentes, pero entonces provocan hipoglucemias graves, potencialmente mortales, o ulceraciones gástricas múltiples.
- Sin embargo, por regla general, los NEN no son funcionales y se diagnostican como hallazgos incidentales o en el contexto de dolencias inespecíficas.
- La única opción potencialmente curativa para las neoplasias neuroendocrinas no metastásicas es la extirpación completa del tumor. A diferencia de otras entidades tumorales, no hay indicación de terapia adyuvante en los tumores neuroendocrinos.
- Las opciones de tratamiento del VEN metastásico han mejorado considerablemente. Para los tumores que expresan SSTR2, se ha establecido el uso de análogos de la somatostatina. Las neoplasias neuroendocrinas de grado superior se tratan con una combinación de platino y etopósido análoga a la terapia del carcinoma bronquial de células pequeñas.
Literatura:
- Cives M, et al: CA Cancer J Clin 2018; 68: 471-487.
- Modlin IM, et al: Endocr Relat Cancer 2014; 21(4): 615-628.
- Deppen A, et al: J Nucl Med 2016; 57(6): 872-878
- Kayani I, et al.: Cáncer 2008; 112(11): 2447-2455
- Delle Fave G, et al: Neuroendocrinología 2016; 103(2): 119-124
- Pape UF, et al: Neuroendocrinología 2016; 103(2): 144-152
- Rinke A, et al: J Clin Oncol 2009; 27(28): 4656-4663.
- Caplin ME, et al: N Engl J Med 2014; 371(3): 224-233.
- Strosberg J, et al: N Engl J Med 2017; 376(2): 125-135
- Yao J, et al: Lancet 2016; 387: 968-977
- Strosberg JR, et al: Cáncer 2011; 117(2): 268-275
InFo ONcOLOGíA & HEMATOLOGíA 2019; 7(1): 16-18.