Le mésothéliome pleural malin est une tumeur agressive qui se développe à partir de la plèvre. Les patients ont une courte espérance de vie après le diagnostic, notamment en raison des possibilités limitées de traitement. L’exposition aux fibres d’amiante est considérée comme le principal facteur de risque pour le développement d’un mésothéliome pleural. Comme la maladie évolue insidieusement sur plusieurs décennies, elle est souvent découverte très tard.
Vous pouvez passer le test de FMC sur notre plateforme d’apprentissage après avoir lu le matériel recommandé. Pour ce faire, veuillez cliquer sur le bouton suivant :
Le mésothéliome pleural malin est une tumeur agressive qui se développe à partir de la plèvre. Les patients ont une courte espérance de vie après le diagnostic, notamment en raison des possibilités limitées de traitement. L’exposition aux fibres d’amiante est considérée comme le principal facteur de risque pour le développement d’un mésothéliome pleural. Comme la maladie évolue insidieusement sur plusieurs décennies, elle n’est souvent détectée que très tard. C’est pourquoi les patients sont souvent diagnostiqués avec un mésothéliome pleural inopérable, qui se trouve déjà à un stade avancé. Cela limite les mesures thérapeutiques et se traduit également par une faible espérance de vie d’environ 12 mois après le diagnostic.
Origine du mésothéliome pleural
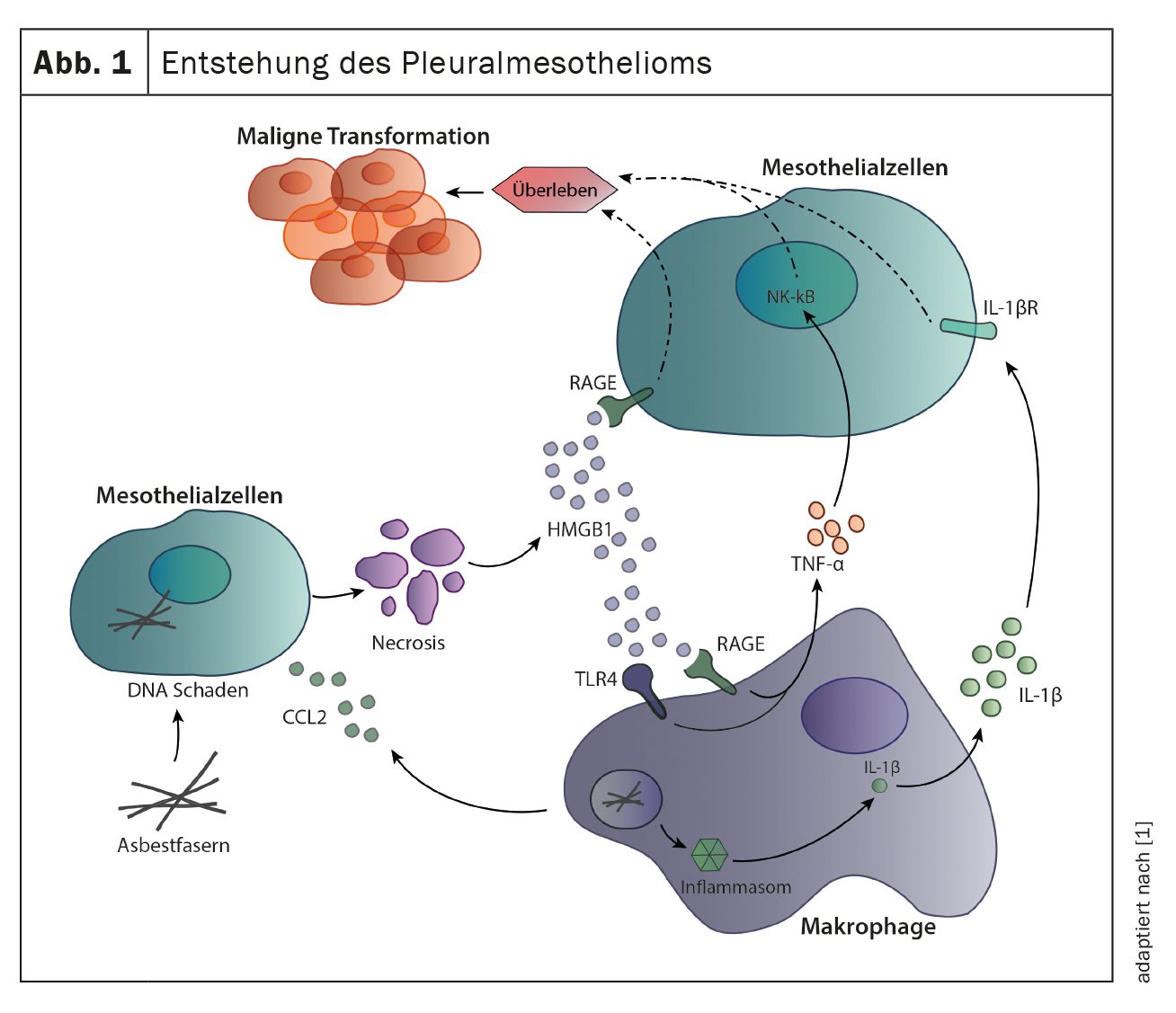
La cause la plus fréquente du développement d’un mésothéliome pleural est l’exposition aux fibres d’amiante, généralement plusieurs décennies avant l’apparition des premiers symptômes. L’inhalation des fibres provoque une inflammation chronique de la plèvre, qui contribue à la transformation maligne des cellules mésothéliales. Les fibres peuvent causer directement des dommages à l’ADN dans les cellules mésothéliales, ce qui entraîne la mort des cellules et la libération de médiateurs inflammatoires tels que HMGB1 et CCL2. Les médiateurs inflammatoires, en particulier CCL2, recrutent des macrophages et agissent directement sur les cellules mésothéliales en se liant aux récepteurs RAGE des cellules mésothéliales et en induisant leur division cellulaire et leur migration. Les macrophages recrutés contribuent également à l’inflammation locale et à la prolifération des cellules mésothéliales. L’ingestion de fibres d’amiante par les macrophages stimule le capteur immunitaire inflammasome, ce qui entraîne la sécrétion d’IL-1β. Outre l’IL-1β, les macrophages sécrètent également le TNF-α, ces deux cytokines contribuant à la survie des cellules mésothéliales et à la poursuite de leur transformation maligne (figure 1) [1].
Signatures génétiques
Environ 80% des tumeurs ont pour origine une exposition à l’amiante survenue plusieurs décennies auparavant. Une prédisposition génétique familiale due à des mutations germinales dans le gène BAP1peut augmenter la probabilité de développer un mésothéliome pleural. Contrairement à d’autres maladies tumorales, dans lesquelles il existe souvent des mutations activatrices dans les oncogènes, le mésothéliome pleural se caractérise principalement par l’altération et la perte de parties entières de chromosomes et par des mutations dans les gènes suppresseurs de tumeurs. Pendant longtemps, peu d’attention a été accordée aux modifications génétiques dans le mésothéliome pleural, en raison du faible taux de mutations de ce type de cancer et de quelques cas où une thérapie ciblée serait possible. Les mutations et délétions les plus fréquentes concernent les gènes BAP1, CDKN2A et NF2. BAP1 est un gène suppresseur de tumeur impliqué dans la réparation des dommages de l’ADN et dans le contrôle du cycle cellulaire. BAP1 est altéré dans environ 45% des mésothéliomes pleuraux, mais ce chiffre peut varier selon les histologies.
CDKN2A est également un gène suppresseur de tumeur qui est souvent supprimé dans le mésothéliome pleural. On trouve des altérations de ce gène dans environ 47% des tumeurs. CDKN2A joue un rôle important dans la régulation du cycle cellulaire, codant pour l’inhibiteur des kinases 4 et 6 dépendantes de la cycline. La perte de ce gène entraîne donc des signaux pro-mitotiques et la survie de la cellule.
NF2 est également impliqué dans la régulation du cycle cellulaire et est modifié dans environ 32% des mésothéliomes pleuraux. NF2 régule la voie de signalisation Hippo via les protéines YAP et TAZ. L’inactivation de NF2 entraîne une hyperactivation de YAP et une division cellulaire incontrôlée.
Seuls quelques mésothéliomes pleuraux présentent des mutations dans des gènes qui peuvent être traités de manière ciblée par des inhibiteurs de petites molécules. Nous avons pu montrer dans une étude que dans environ 5% de tous les mésothéliomes pleuraux, les gènes ALK, KRAS EGFR, PDGFRA/B, ERBB2 ou FGFR3 sont mutés et pourraient être traités par des thérapies ciblées [2]. D’autres études doivent encore démontrer l’efficacité de ce traitement dans le mésothéliome pleural.
Environnement de la tumeur
L’interaction entre les différents types de cellules, telles que les cellules immunitaires, les cellules stromales, les cellules tumorales et les cellules endothéliales des vaisseaux sanguins dans la tumeur est très complexe et n’a pas encore été entièrement étudiée dans le mésothéliome pleural. Selon le type de tumeur et le patient, l’hétérogénéité des types de cellules et des fonctions est grande. En raison de cela, les thérapies qui agissent directement sur l’environnement tumoral sont difficiles à développer. De plus, les cellules immunitaires ont souvent un phénotype inhibiteur et régulateur, principalement représenté par les lymphocytes T régulateurs, les macrophages de type 2 et les cellules suppressives myéloïdes.
Les macrophages sont les cellules immunitaires les plus courantes dans le mésothéliome pleural et sont recrutés dans le sang sous forme de monocytes par CCL2, qui est sécrété par les cellules mésothéliales. Les macrophages associés à la tumeur expriment un phénotype immunosuppresseur de type 2 et soutiennent la prolifération des cellules mésothéliales malignes et la croissance tumorale, ce qui les met également en corrélation avec un mauvais pronostic. Les deuxièmes cellules immunitaires les plus fréquentes dans le mésothéliome pleural sont les lymphocytes T, dont tous les sous-types sont présents, comme les cellules T auxiliaires CD4+, les cellules T cytotoxiques CD8+ et les cellules T régulatrices FoxP3+. Les cellules T cytotoxiques expriment souvent des marqueurs tels que Lag-3, Tim-3, PD-1, qui définissent un phénotype non réactif. Ces cellules T ne sont plus capables d’exercer des fonctions effectrices, ce qui offre un avantage de survie aux cellules tumorales. Dans le mésothéliome pleural, la présence de cellules T dans la tumeur est positivement associée à la survie, selon les études, mais cela peut varier en fonction de l’histologie et du phénotype spécifique des cellules T. Ainsi, les cellules T régulatrices dans la tumeur sont associées à une survie plus courte. D’autres cellules immunitaires suppressives dans la tumeur sont les cellules suppressives myéloïdes, qui peuvent représenter jusqu’à 10% de toutes les cellules immunitaires infiltrantes. Elles ont un effet négatif sur les cellules T et peuvent inhiber leur division cellulaire.
Pathologie
Le mésothéliome pleural est divisé en trois sous-types histologiques : le mésothéliome pleural épithélioïde (environ 80% des cas), le mésothéliome pleural biphasique et le mésothéliome pleural sarcomatoïde. Le sous-type biphasique se caractérise par une combinaison de structures épithélioïdes et sarcomatoïdes.
Les sous-types histologiques se distinguent principalement par leur espérance de vie. Les patients atteints de mésothéliome pleural épithélioïde ont une espérance de vie plus longue que les patients atteints de mésothéliome pleural biphasique ou sarcomatoïde. De plus, les patients présentant un sous-type épithélioïde bénéficient généralement d’une résection, alors que les autres sous-types ne tirent aucun avantage d’une intervention chirurgicale. Il est parfois difficile de diagnostiquer un mésothéliome pleural sur la base de la morphologie cellulaire, car la plèvre est souvent altérée par des modifications inflammatoires ou en présence de métastases d’une autre maladie maligne. C’est pourquoi il est nécessaire de poursuivre l’analyse d’une biopsie pleurale par immunohistochimie (IHC) de deux marqueurs de mésothéliome tels que la calrétinine, la podoplanine, la tumeur de Wilms 1 (WT-1) ou la cytokératine 5/6. En outre, d’autres carcinomes peuvent être exclus par la coloration avec l’ACE, le Ber-EP4, la pancytokératine ou la claudine-4. Un mésothéliome pleural avec une modification de type épithélial peut être distingué d’un carcinome épidermoïde par une coloration pour les marqueurs p40 et p63. En outre, les altérations génétiques des gènes BAP1 et CDKN2A sont fréquentes, ce qui entraîne la perte de l’expression de ces protéines dans la tumeur. Par conséquent, une analyse immunohistochimique de l’expression de BAP1 et de MTAP (MTAP est analysé comme substitut de CDKN2A, car ces gènes sont situés côte à côte sur le segment chromosomique 9p21 et il y a souvent une co-délétion) peut également être utile pour le diagnostic final.
Symptômes, diagnostic et stadification
Les patients se présentent souvent avec des symptômes peu clairs de dyspnée, de douleur thoracique et de perte de poids. Souvent, les patients présentent un épanchement pleural unilatéral. Le diagnostic de mésothéliome pleural repose sur plusieurs examens : i) des examens radiologiques, y compris un scanner du thorax, ii) une biopsie pleurale par thoracoscopie pour vérifier le diagnostic de mésothéliome pleural et déterminer l’histologie. La biopsie pleurale doit donc toujours s’étendre à la graisse sous-pleurale et être réalisée à partir de trois sites différents ou plus. Pour éviter le risque d’implantation de cellules tumorales dans la paroi thoracique, seuls 1 ou 2 sites d’entrée de la thoracoscopie devraient être utilisés [3]. Il est préférable de les placer dans le même espace intercostal de la résection macroscopiquement complète prévue dans la suite du traitement.
Le staging se fait par tomographie par émission de positons (PET-CT) et est complété par une médiastinoscopie ou une échographie endobronchique (EBUS) en cas de suspicion d’atteinte des ganglions lymphatiques médiastinaux, par une thoracoscopie controlatérale en cas de suspicion d’atteinte de la plèvre controlatérale, ou par une laparoscopie en cas de suspicion d’atteinte du péritoine. L’imagerie par résonance magnétique (IRM) du thorax peut en outre fournir des informations précieuses pour le staging en cas d’infiltration dans le diaphragme, la paroi thoracique, le péricarde ou le médiastin.
Interventions chirurgicales
Le traitement doit être discuté dans le cadre d’un Tubmorboard interdisciplinaire réunissant des spécialistes de la chirurgie thoracique, de l’oncologie, de la radio-oncologie, de la pathologie et de la radiologie. Si le stade de la tumeur et l’état général du patient se prêtent à un concept de traitement multimodal, une chimiothérapie néoadjuvante avec des cytostatiques à base de platine et des antagonistes de l’acide folique est suivie d’un re-positionnement par PET-CT pour réévaluer l’opérabilité. En raison de la situation anatomique avec proximité des structures médiastinales, il n’est pas possible de respecter des distances de sécurité suffisantes lors d’une résection du MPM. Une résection radicale signifie donc ici une résection macroscopiquement complète avec pour objectif une cytoréduction maximale, mais avec le risque d’une tumeur microscopique résiduelle [4]. Une résection macroscopiquement complète peut être obtenue par une pneumonectomie extrapleurale (EPP) ou une pleurectomie élargie et décortication (EPD) préservant le parenchyme pulmonaire [4]. Alors que l’EPP consiste en une résection en bloc du poumon atteint avec la plèvre viscérale et pariétale, ainsi que le diaphragme et le péricarde, l’EPD consiste uniquement à détacher la plèvre pariétale et viscérale et à l’enlever avec le diaphragme et le péricarde atteints, tout en conservant le poumon [4]. En l’absence de signes d’atteinte péricardique ou diaphragmatique, une pleurectomie et une décortication (DP) isolées peuvent être choisies. Pour toutes les résections chirurgicales, une lymphadénectomie médiastinale systématique doit également être effectuée. Ces dernières années, on assiste à un glissement progressif de l’EPP vers l’EPD, car le parenchyme pulmonaire et les réserves fonctionnelles sont préservés, ce qui permet au patient d’avoir une meilleure qualité de vie. En outre, l’EPP est associée à une augmentation de la morbidité et de la mortalité périopératoires. L’EPP ne devrait être envisagée que dans des cas sélectionnés, avec une infiltration étendue du parenchyme pulmonaire et des réserves cardiopulmonaires suffisantes, et ne devrait être réalisée que dans des centres expérimentés.
Chez les patients pour lesquels une cytoréduction maximale par résection macroscopique complète n’est pas envisageable, la réapparition d’un épanchement pleural symptomatique peut être évitée dans une approche palliative. En cas de poumon étendu, on procède à une pleurodèse de suif par thoracoscopie, et en cas de poumon chroniquement piégé, on utilise un système de cathéter tunnelisé sous-cutané qui permet de drainer régulièrement l’épanchement, même à domicile.
Une VATS-PP palliative est recommandée pour contrôler les épanchements pleuraux répétés chez les patients qui sont suffisamment aptes pour un traitement chirurgical et qui ne peuvent pas bénéficier d’une pleurodèse chimique (ou après une pleurodèse infructueuse) ou d’un cathéter à demeure [5].
Thérapie systémique
Depuis 2004, les patients sont traités par un traitement systémique associant le pemetrexed et une chimiothérapie à base de platine. L’introduction du bevacizumab, un inhibiteur de l’angiogenèse, en association avec le cisplatine/pemetrexed a permis d’augmenter l’espérance de vie d’environ 2,5 mois. Suite au succès des inhibiteurs de points de contrôle immunitaires dans différentes tumeurs solides, le traitement combiné d’ipilimumab (anticorps anti-CTLA-4) et de nivolumab (anticorps anti-PD-1) a été testé. Ce traitement a montré une amélioration significative de la survie dans le mésothéliome pleural sarcomatoïde et épithélioïde avec PD-L1>1% [6]. Par conséquent, ce traitement a été approuvé par la FDA et l’EMA comme traitement de première ligne en 2020. Cependant, dans les lignes de traitement ultérieures, le mésothéliome pleural reste toujours une maladie sans traitement standardisé et les patients sont souvent inclus dans des essais cliniques.
Messages Take-Home
- Une cytoréduction chirurgicale maximale par résection macroscopique complète est recommandée chez des patients sélectionnés présentant un MPM à un stade précoce et disposant de réserves cardiopulmonaires suffisantes.
- Une résection macroscopiquement complète doit toujours être réalisée dans le cadre d’un concept thérapeutique multimodal en combinaison avec une chimiothérapie.
- Les recommandations et les décisions de traitement devraient toujours être prises lors d’un tumorboard interdisciplinaire d’oncologie thoracique en présence d’oncologues, de radio-oncologues, de pneumologues et de chirurgiens thoraciques.
- Le traitement de première ligne chez les patients non opérables est basé sur le nivolumab et l’ipilimumab pour les patients atteints de mésothéliome pleural épithélioïde avec expression de PD-L1 >1% et pour tous les autres sous-types histologiques. Sinon, l’autre option est un traitement par platinum-pemetrexed et bevacizumab.
Littérature :
- Hiltbrunner S, Mannarino L, Kirschner MB, et al.: Tumor Immune Microenvironment and Genetic Alterations in Mesothelioma, Frontiers in oncology 11 (2021) 660039.
- Hiltbrunner S, Fleischmann Z, Sokol E, Curioni-Fontecedro A: 1734P Genomic landscape of pleural and peritoneal mesothelioma tumors, Annals of Oncology 32 (2021) S1200.
- Kindler HL, Ismaila N, Armato SG, et al.: Treatment of Malignant Pleural Mesothelioma: American Society of Clinical Oncology Clinical Practice Guideline, J Clin Oncol 36(13) (2018): 1343–1373.
- Rice D, Rusch V, Pass H, et al.: Recommendations for uniform definitions of surgical techniques for malignant pleural mesothelioma: a consensus report of the international association for the study of lung cancer international staging committee and the international mesothelioma interest group, J Thorac Oncol 6(8) (2011): 1304–1312.
- Opitz I, Scherpereel A, Berghmans T, et al.: ERS/ESTS/EACTS/ESTRO guidelines for the management of malignant pleural mesothelioma, Eur J Cardiothorac Surg 58(1) (2020): 1–24.
- Baas P, Scherpereel A, Nowak AK, et al. : First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743) : a multicentre, randomised, open-label, phase 3 trial, Lancet 397(10272) (2021) 375-386.
InFo ONKOLOGIE & HÄMATOLOGIE 2024; 12(6): 6–9