O mesotelioma pleural maligno é um tumor agressivo com origem na pleura. Os pacientes têm uma esperança de vida curta após o diagnóstico, também devido às opções de tratamento limitadas. A exposição às fibras de amianto é considerada o principal factor de risco para o desenvolvimento de mesotelioma pleural. Uma vez que a doença progride de forma insidiosa ao longo de décadas, é frequentemente descoberta muito tarde.
Pode fazer o teste CME na nossa plataforma de aprendizagem depois de rever os materiais recomendados. Clique no botão seguinte:
O mesotelioma pleural maligno é um tumor agressivo com origem na pleura. Os pacientes têm uma esperança de vida curta após o diagnóstico, também devido às opções de tratamento limitadas. A exposição às fibras de amianto é considerada o principal factor de risco para o desenvolvimento de mesotelioma pleural. Uma vez que a doença progride de forma insidiosa ao longo de décadas, é frequentemente descoberta muito tarde. É por esta razão que os doentes são frequentemente diagnosticados com mesotelioma pleural inoperável, que já se encontra numa fase avançada. Isto limita as medidas terapêuticas e reflecte-se também numa baixa esperança de vida de cerca de 12 meses após o diagnóstico.
Desenvolvimento de mesotelioma pleural
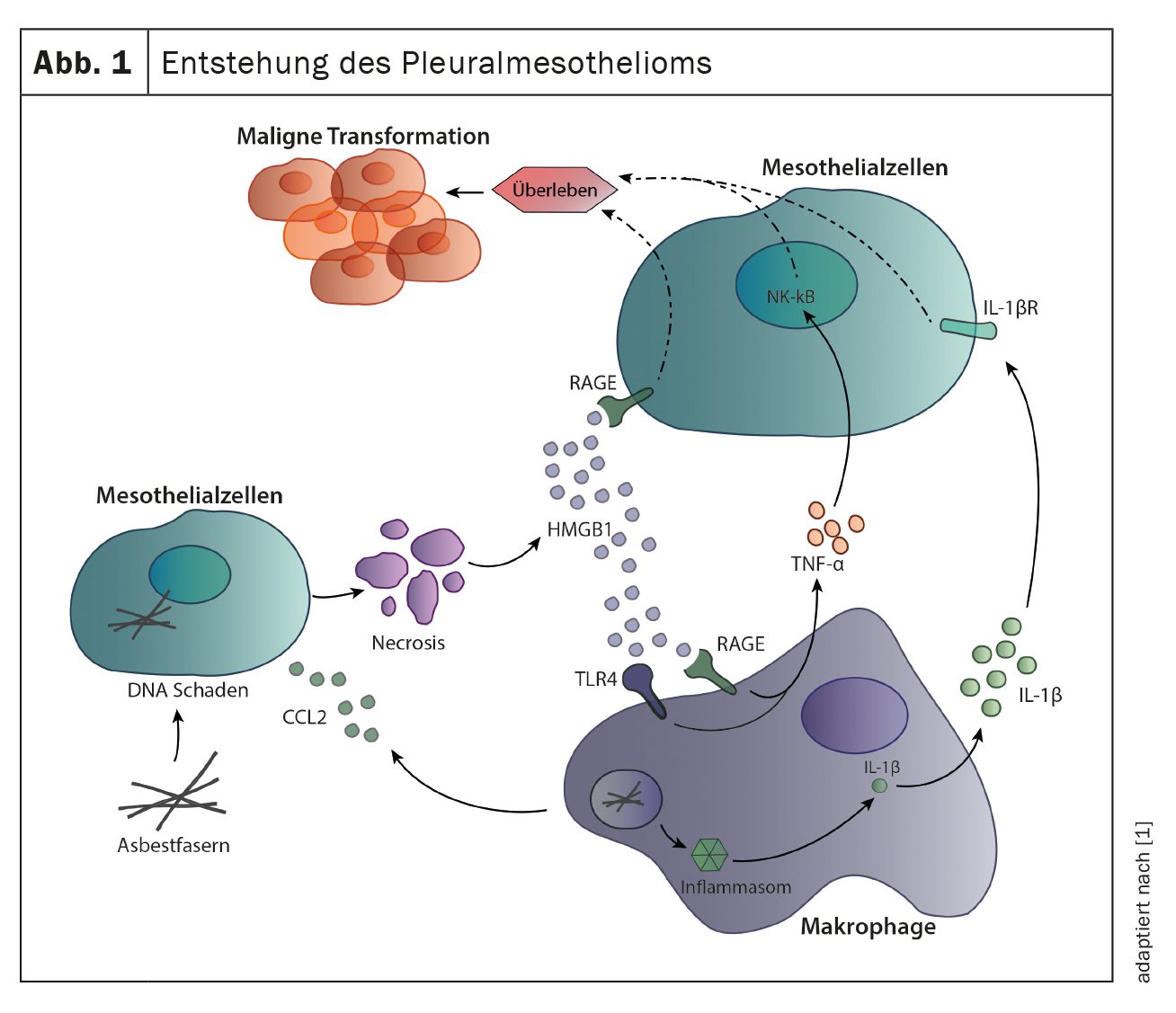
A causa mais comum para o desenvolvimento do mesotelioma pleural é a exposição às fibras de amianto, geralmente décadas antes do aparecimento dos primeiros sintomas. A inalação das fibras leva à inflamação crónica da pleura, o que contribui para a transformação maligna das células mesoteliais. As fibras podem causar directamente danos de ADN em células mesoteliais, levando à morte celular e à libertação de mediadores inflamatórios tais como HMGB1 e CCL2. Os mediadores inflamatórios, especialmente o CCL2, recrutam macrófagos e actuam directamente sobre células mesoteliais ligando-se aos receptores RAGE em células mesoteliais e induzindo a sua divisão e migração celular. Os macrófagos recrutados também contribuem para a inflamação local e proliferação de células mesoteliais. A absorção das fibras de amianto pelos macrófagos estimula o inflamassoma do sensor imunitário, que leva à secreção de IL-1β. Para além da IL-1β, os macrófagos também segregam TNF-α, contribuindo ambas as citocinas para a sobrevivência das células mesoteliais e para a sua posterior transformação maligna (Fig. 1) [1].
Assinaturas Gene
Cerca de 80% dos tumores têm uma exposição ao amianto décadas antes. Uma predisposição genética familiar devida a mutações na linha germinal do gene BAP1pode aumentar a probabilidade de desenvolvimento de mesotelioma pleural. Em contraste com outras doenças tumorais, nas quais estão frequentemente presentes mutações activadoras em oncogenes, o mesotelioma pleural caracteriza-se principalmente pela alteração e perda de partes cromossómicas inteiras e por mutações nos genes supressores de tumores. Durante muito tempo, foi dada pouca atenção às alterações genéticas no mesotelioma pleural, devido à baixa taxa de mutação deste tipo de cancro e aos poucos casos em que seria possível uma terapia dirigida. As mutações e supressões mais comuns afectam os genes BAP1, CDKN2A e NF2. BAP1 é um gene supressor de tumores envolvido na reparação de danos no ADN e no controlo do ciclo celular. O BAP1 está alterado em cerca de 45% de todos os mesoteliomas pleurais, embora este valor possa variar consoante as diferentes histologias.
O CDKN2A é também um gene supressor de tumores que é frequentemente eliminado no mesotelioma pleural. As alterações neste gene encontram-se em cerca de 47% de todos os tumores. O CDKN2A desempenha um papel importante na regulação do ciclo celular, codificando o inibidor da quinase dependente de ciclina 4 e 6. A perda deste gene conduz, portanto, à sinalização pró-mitótica e à sobrevivência celular.
O NF2 também está envolvido na regulação do ciclo celular e está alterado em cerca de 32% de todos os mesoteliomas pleurais. NF2 regula o caminho de sinalização Hipopótamo através das proteínas YAP e TAZ. A inativação de NF2 leva à hiperactivação de YAP e a uma divisão celular descontrolada.
Apenas alguns mesoteliomas pleurais têm mutações em genes que podem ser alvo de inibidores de pequenas moléculas. Conseguimos mostrar num estudo que em cerca de 5% de todos os mesoteliomas pleurais os genes ALK, KRAS EGFR, PDGFRA/B, ERBBB2 ou FGFR3 são mutantes, que poderiam ser tratados com terapias específicas [2]. Outros estudos têm ainda de mostrar a eficácia deste tratamento no mesotelioma pleural.
Ambiente tumoral
A interacção de diferentes tipos de células, tais como células imunitárias, células do estroma, células tumorais e células endoteliais dos vasos sanguíneos do tumor é muito complexa e ainda não totalmente compreendida no mesotelioma pleural. Dependendo do tipo de tumor e do paciente, a heterogeneidade de tipos de células e funções é grande. Devido a isto, as terapias que actuam directamente sobre o ambiente tumoral são difíceis de desenvolver. Além disso, as células imunitárias têm frequentemente um fenótipo inibitório e regulador, representado principalmente por células T reguladoras, macrófagos tipo 2 e células supressoras de mielóide.
Os macrófagos são as células imunitárias mais comuns no mesotelioma pleural e são recrutados a partir do sangue sob a forma de monócitos pelo CCL2, que é secretado pelas células mesoteliais. Os macrófagos associados ao tumor exprimem um fenótipo imunossupressor do tipo 2 e apoiam a proliferação de células mesoteliais malignas e o crescimento de tumores, correlacionando-se também com um mau prognóstico. As segundas células imunes mais abundantes no mesotelioma pleural são os linfócitos T, com todos os subtipos presentes como as células T helper CD4+, as células T citotóxicas CD8+ e as células T reguladoras FoxP3+. As células T citotóxicas expressam frequentemente marcadores tais como Lag-3, Tim-3, PD-1, que definem um fenótipo não reactivo. Estas células T já não são capazes de desempenhar funções efectoras, o que dá às células tumorais uma vantagem de sobrevivência. A presença de células T no tumor está positivamente associada à sobrevivência no mesotelioma pleural, dependendo do estudo, mas isto pode variar dependendo da histologia e do fenótipo específico das células T. Assim, as células T reguladoras do tumor estão associadas a uma sobrevivência mais curta. Outras células imunitárias supressoras do tumor são as células supressoras de mielóide, que podem representar até 10% de todas as células imunitárias infiltrantes. Têm um efeito negativo sobre as células T e podem inibir a sua divisão celular.
Patologia
O mesotelioma pleural está dividido em três subtipos histológicos, epitélioide (aproximadamente 80% dos casos), mesotelioma pleural bifásico e sarcomatóide. O subtipo bifásico é caracterizado por uma combinação de estruturas epitelioides e sarcomatoides.
Os subtipos histológicos diferem principalmente em termos de esperança de vida. Os doentes com mesotelioma pleural epitelióide têm uma maior esperança de vida em comparação com os doentes com mesotelioma pleural bifásico ou sarcomatóide. Além disso, os pacientes com um subtipo de epitélioide beneficiam geralmente da ressecção, enquanto que os outros subtipos não beneficiam da cirurgia. Por vezes o diagnóstico de mesotelioma pleural é difícil devido à morfologia celular, porque por um lado a pleura é frequentemente alterada por alterações inflamatórias ou estão presentes metástases de outra doença maligna. Portanto, é necessária uma análise mais aprofundada de uma biópsia pleural por imuno-histoquímica (IHC) de dois marcadores de mesotelioma tais como calretinina, podoplanina, tumor de Wilms-1 (WT-1) ou citoqueratina 5/6. Além disso, outros carcinomas podem ser excluídos por coloração com CEA, Ber-EP4, pancitoceratina ou claudina-4. O mesotelioma pleural com uma mudança tipo célula escamosa pode ser distinguido do carcinoma de células escamosas por coloração para os marcadores p40 e p63. Além disso, ocorrem frequentemente alterações genéticas nos genes BAP1 e CDKN2A, resultando na perda de expressão destas proteínas no tumor. Devido a isto, a análise imuno-histoquímica da expressão BAP1 e MTAP (o MTAP é analisado como um proxy para CDKN2A, uma vez que estes genes estão localizados directamente adjacentes uns aos outros no segmento cromossómico 9p21 e a co-deleção está frequentemente presente) também pode ser útil para fazer um diagnóstico definitivo.
Sintomas, diagnóstico e encenação
Os pacientes apresentam frequentemente sintomas inexplicáveis de dispneia, dores no peito e perda de peso. Os doentes apresentam frequentemente uma efusão pleural unilateral. O diagnóstico do mesotelioma pleural é feito através de vários exames: i) Exames radiológicos, incluindo a TAC do tórax, ii) Biópsia pleural por toracoscopia para confirmar o diagnóstico de mesotelioma pleural e determinar a histologia. Por conseguinte, a biopsia pleural deve sempre estender-se à gordura subpleural e ser efectuada em três ou mais localizações diferentes. Para prevenir o risco de implantação de células tumorais na parede torácica, só devem ser utilizados 1-2 locais de entrada toracoscópica [3]. De preferência, estes são colocados no mesmo espaço intercostal da ressecção macroscopicamente completa planeada no curso seguinte.
O estadiamento é feito por tomografia por emissão de positrões (PET-CT) e é suplementado por mediastinoscopia ou ultra-som endobrônquico (EBUS) se se suspeitar de envolvimento de gânglios linfáticos mediastinais, por toracoscopia contralateral se se suspeitar de envolvimento pleural contralateral, ou por laparoscopia se se suspeitar de envolvimento peritoneal. A ressonância magnética (RM) do tórax também pode fornecer informação valiosa para fins de encenação sobre infiltração no diafragma, parede torácica, pericárdio ou mediastino.
Intervenções cirúrgicas
O tratamento deve ser discutido num tubmorboard interdisciplinar com representantes especializados em cirurgia torácica, oncologia, radiooncologia, patologia e radiologia. Se a fase do tumor e o estado geral do paciente o qualificam para um conceito de tratamento multimodal, a quimioterapia neoadjuvante com citostáticos contendo platina e antagonistas do ácido fólico é seguida de uma nova fase usando PET-CT para reavaliar a operabilidade. Devido à situação anatómica com proximidade de estruturas mediastinais, não podem ser mantidas distâncias de segurança suficientes durante uma ressecção MPM. A ressecção radical aqui significa, portanto, uma ressecção macroscópica completa com o objectivo de máxima citoredução, mas com o risco de tumor microscópico residual [4]. Uma ressecção macroscopicamente completa pode ser conseguida através de uma pneumonectomia extrapleural (PPE) ou de uma pleurectomia alargada e decorticação (EPD) preservadora do parênquima pulmonar [4]. Enquanto o PPE envolve uma ressecção em bloco do pulmão afectado com pleura visceral e parietal, bem como diafragma e pericárdio, na EPD apenas a pleura parietal e visceral é descolada e removida juntamente com o diafragma e pericárdio afectados, deixando o pulmão intacto [4]. Na ausência de sinais de envolvimento pericárdico ou diafragmático, a pleurectomia isolada e a decorticação (PD) podem ser eleitas. Em todas as ressecções cirúrgicas, deve também ser realizada uma linfadenectomia mediastinal sistemática. Nos últimos anos, tem havido uma mudança crescente do PPE para a EPD, à medida que o parênquima pulmonar e as reservas funcionais são preservados, permitindo que o paciente tenha uma melhor qualidade de vida. O EPP está também associado ao aumento da morbilidade e mortalidade perioperatória. A PPE só deve ser considerada em casos selecionados com infiltração extensa do parênquima pulmonar e reservas cardiopulmonares suficientes e só deve ser realizada em centros experientes.
Em doentes em que a citoredução máxima por ressecção macroscópica completa não é uma opção, a recorrência de derrame pleural sintomático pode ser evitada numa abordagem paliativa. No caso de pulmões extensos, isto é feito através de pleurodese toracoscópica com talco e, no caso de pulmões cronicamente presos, através de um sistema de cateter subcutâneo, que também pode ser utilizado para a drenagem regular de efusões no ambiente doméstico.
O VATS-PP paliativo é recomendado para controlar efusões pleurais recorrentes em pacientes que estão suficientemente aptos para o tratamento cirúrgico e não podem beneficiar da pleurodese química (ou após uma pleurodese mal sucedida) ou de cateteres residentes [5].
Terapia do sistema
Desde 2004, os pacientes têm sido tratados com uma terapia de combinação sistémica de quimioterapia de base pemetrexada e platina. A introdução de bevacizumab, um inibidor da angiogénese, em combinação com cisplatina/pemetrexed foi capaz de aumentar a esperança de vida em cerca de 2,5 meses. Com base no sucesso dos inibidores do ponto de controlo imunitário em vários tumores sólidos, foi testada a terapia combinada de ipilimumab (anticorpo anti-CTLA-4) e nivolumab (anticorpo anti-PD-1). Esta terapia mostrou uma melhoria significativa na sobrevivência do mesotelioma pleural sarcomatóide e epithelioide com PD-L1>1% [6]. Assim, este tratamento foi aprovado pela FDA e pela EMA como terapia de primeira linha em 2020. Contudo, em linhas de tratamento posteriores, o mesotelioma pleural continua a ser uma doença sem tratamentos padronizados e os pacientes são frequentemente inscritos em ensaios clínicos.
Mensagens para levar para casa
- Em doentes selecionados com MPM em fase inicial e reservas cardiopulmonares suficientes, recomenda-se a citorredução cirúrgica máxima através de uma ressecção macroscópica completa.
- Uma ressecção macroscopicamente completa deve ser sempre efectuada como parte de um conceito de terapia multimodal em combinação com quimioterapia.
- As recomendações e decisões de tratamento devem ser sempre tomadas num comité interdisciplinar de tumores de oncologia torácica, na presença de oncologistas, radiologistas, pneumologistas e cirurgiões torácicos.
- A terapia de primeira linha em pacientes não operáveis baseia-se no nivolumab e ipilimumab para pacientes com mesotelioma pleural epitelioide com expressão PD-L1 >1% e para todos os outros subtipos histológicos. Caso contrário, o tratamento com platina-pemetrexedo e bevacizumab é a outra opção.
Literatura:
- Hiltbrunner S, Mannarino L, Kirschner MB, et al.: Tumor Immune Microenvironment and Genetic Alterations in Mesothelioma, Frontiers in oncology 11 (2021) 660039.
- Hiltbrunner S, Fleischmann Z, Sokol E, Curioni-Fontecedro A: 1734P Genomic landscape of pleural and peritoneal mesothelioma tumors, Annals of Oncology 32 (2021) S1200.
- Kindler HL, Ismaila N, Armato SG, et al.: Treatment of Malignant Pleural Mesothelioma: American Society of Clinical Oncology Clinical Practice Guideline, J Clin Oncol 36(13) (2018): 1343–1373.
- Rice D, Rusch V, Pass H, et al.: Recommendations for uniform definitions of surgical techniques for malignant pleural mesothelioma: a consensus report of the international association for the study of lung cancer international staging committee and the international mesothelioma interest group, J Thorac Oncol 6(8) (2011): 1304–1312.
- Opitz I, Scherpereel A, Berghmans T, et al.: ERS/ESTS/EACTS/ESTRO guidelines for the management of malignant pleural mesothelioma, Eur J Cardiothorac Surg 58(1) (2020): 1–24.
- Baas P, Scherpereel A, Nowak AK, et al.: First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial, Lancet 397(10272) (2021) 375–386.
InFo ONKOLOGIE & HÄMATOLOGIE 2024; 12(6): 6–9