Il existe un grand nombre de mutations différentes qui peuvent être à l’origine d’une dystrophie rétinienne héréditaire. Grâce au développement de méthodes modernes d’analyse génétique moléculaire, la plupart des causes génétiques peuvent aujourd’hui être identifiées. Un médicament de thérapie génique innovant est disponible pour les patients atteints de dystrophie rétinienne due à des mutations bialléliques du RPE65.
Le terme générique de “dystrophies rétiniennes héréditaires” (IRD ; inherited retinal diseases) regroupe un ensemble hétérogène de maladies aux phénotypes qui se chevauchent et qui se caractérisent par une dégénérescence et un dysfonctionnement progressifs de la rétine [1]. Le point commun de ces maladies est qu’elles affectent l’ensemble de la rétine au cours de leur évolution et que la vision se détériore lentement et progressivement au cours de la vie. Les IRD peuvent être causées par des mutations dans >250 gènes différents [2]. Tant la pathogenèse moléculaire que le tableau clinique et l’évolution de la maladie sont définis de manière déterminante par le type et la nature du défaut génétique [3]. En fonction du gène muté, différents troubles du développement et du fonctionnement de la rétine peuvent survenir, comme par exemple des troubles du métabolisme rétinien, des structures cellulaires ou de la phototransduction. La forme la plus courante d’IRD est la rétinite pigmentaire, à laquelle s’ajoutent l’amaurose congénitale de Leber et la dystrophie rétinienne à début précoce (EORD ; early-onset retinal dystrophy) [2].
Un diagnostic précoce est essentiel
La rétinite pigmentaire se caractérise par une dégénérescence des bâtonnets au stade initial de la maladie, ce qui se traduit par une cécité nocturne. Les symptômes caractéristiques de l’amaurose congénitale de Leber comprennent une diminution drastique de l’acuité visuelle et des pertes du champ visuel. Un diagnostic précoce et correct de la maladie est très important, tout comme une description morphologique et fonctionnelle approfondie de l’état actuel de la rétine en fonction des mutations sous-jacentes dans le gène RPE65. En effet, l’évolution rapide et grave de la maladie rend nécessaire une intervention précoce tant que les cellules de l’épithélium pigmentaire rétinien, qui sont les cellules cibles, sont encore présentes, de même que les photorécepteurs. Dans le cadre du diagnostic fonctionnel ophtalmologique et en particulier du diagnostic par imagerie non invasive (par ex. tomographie par cohérence optique, autofluorescence du fond d’œil ou proche infrarouge), la description clinique du phénotype est possible [5,6]. Pour identifier la cause génétique des dystrophies rétiniennes héréditaires, il est indispensable de recourir à des techniques de génétique moléculaire telles que le séquençage de nouvelle génération.
L’imagerie par autofluorescence fournit des résultats importants
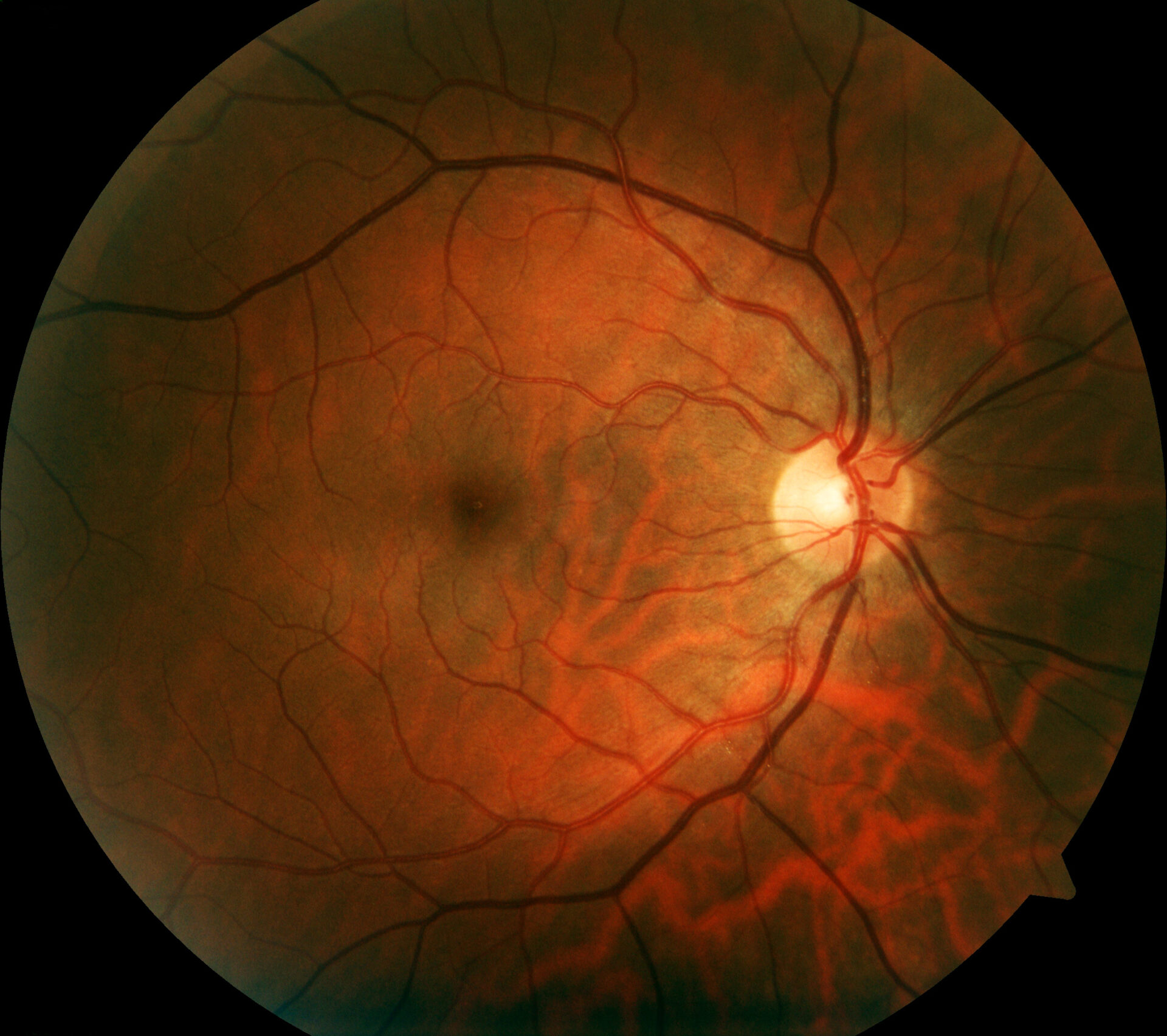
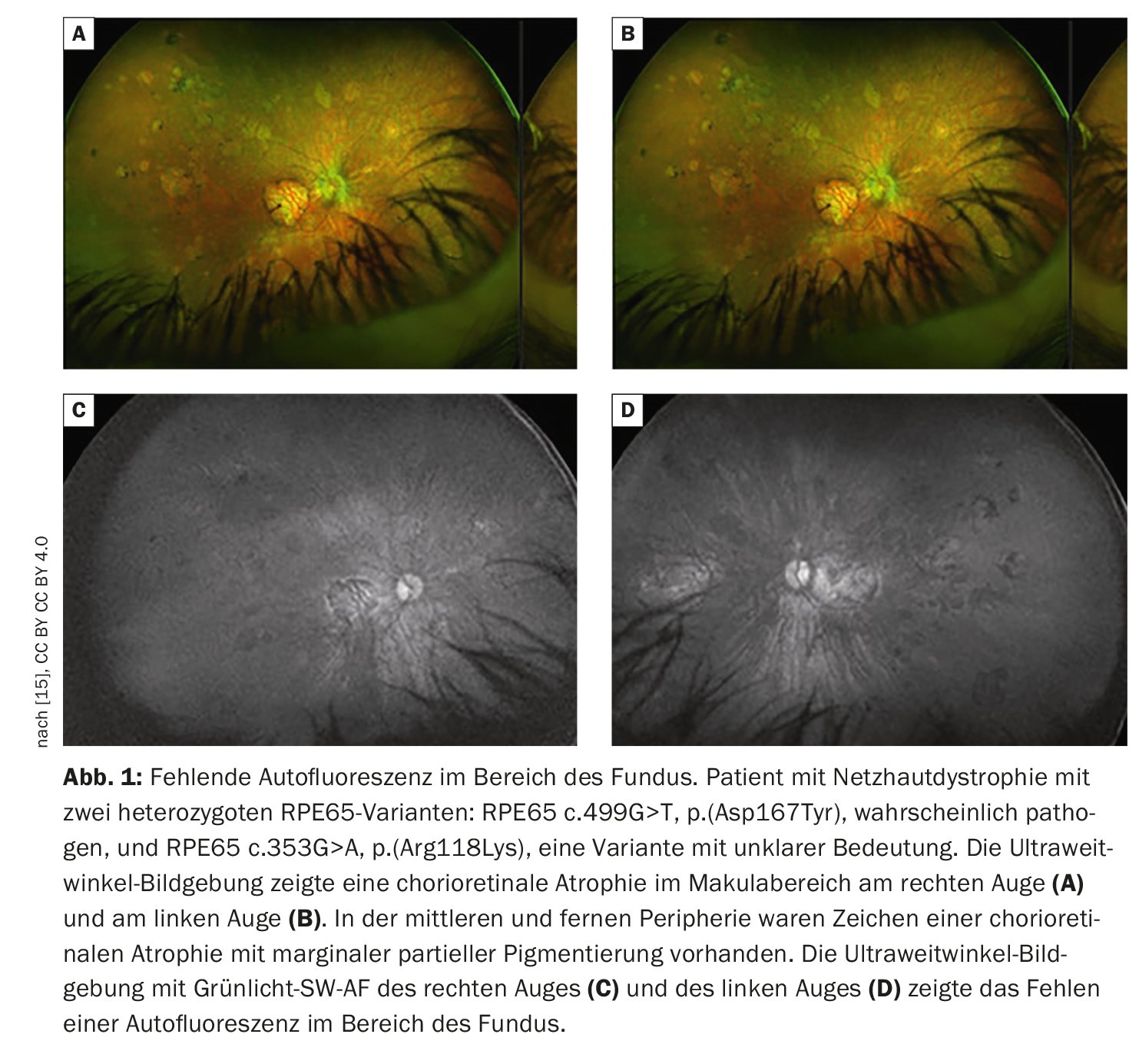
L’imagerie rétinienne permet de visualiser les pathologies rétiniennes de manière plus détaillée que l’examen fonduscopique. Outre la photographie conventionnelle du fond d’œil en couleur, la tomographie par cohérence optique (OCT) à haute résolution et l’autofluorescence du fond d’œil, qui permet de visualiser les fluorophores du fond d’œil au moyen d’une lumière à ondes courtes, sont des méthodes bien établies. L’autofluorescence du fond de l’œil permet de visualiser la répartition des fluorophores du fond de l’œil, en utilisant généralement une lumière d’excitation à ondes courtes dans la gamme du bleu ou du vert. Chez les patients présentant des mutations dans le gène RPE65, l’absence ou du moins la diminution de l’autofluorescence dans la région du fond d’œil (Fig. 1) est une caractéristique cliniquement significative, due à un métabolisme défectueux des rétinoïdes dans les photorécepteurs et les cellules RPE [9]. Par exemple, la rétinite pigmentaire se caractérise par un anneau concentrique d’autofluorescence élevée pour lequel il n’existe pas de corrélation visible à la loupe [10–12]. Bien que l’origine exacte de ce phénomène soit incomplètement comprise à ce jour, il a été démontré par des études OCT que l’anneau correspond à la perte de la bande ellipsoïdale et à un amincissement important, voire à la perte de la couche de photorécepteurs [10].
Test de génétique moléculaire pour la détection de la cause génétique
Les phénotypes des variants de différents gènes se chevauchent souvent et une variabilité considérable est possible pour les variants d’un seul gène. C’est pourquoi un test de génétique moléculaire est indispensable et constitue une base importante pour le choix de l’option thérapeutique appropriée [6]. Dans 55 à 80% des cas, la cause génétique des dystrophies rétiniennes héréditaires peut être identifiée par des analyses de génétique moléculaire [4]. Chez les patients présentant des mutations bialléliques du gène RPE65, l’isomérase du RPE65 n’est pas exprimée dans l’épithélium pigmentaire rétinien (EPR) ou est exprimée sous forme de protéine non fonctionnelle. Par définition, pour diagnostiquer une dystrophie rétinienne associée à une mutation biallélique du RPE65, les deux copies du gène RPE65 doivent être présentes sous la forme d’une ou de deux variantes pathogènes différentes [8]. Le phénotype clinique dans lequel se manifestent les mutations bialléliques du RPE65 est généralement attribué à l’amaurose congénitale de Leber ou à la rétinite pigmentaire. Un déficit en RPE65 isomérase fonctionnelle a pour conséquence que le tout-trans-rétinal produit dans le cadre de la cascade de phototransduction n’est pas converti en 11-cis-rétinal, ce qui perturbe la régénération du pigment visuel rhodopsine [1]. Cette perturbation du métabolisme rétinien fait perdre aux photorécepteurs leur capacité à réagir aux stimuli lumineux. De plus, l’accumulation d’intermédiaires toxiques entraîne la mort des cellules épithéliales pigmentaires rétiniennes, ce qui provoque un dysfonctionnement supplémentaire des photorécepteurs. Cet effet est plus prononcé pour les bâtonnets de photorécepteurs que pour les cônes de photorécepteurs, car ces derniers présentent encore une voie métabolique alternative pour la régénération du pigment visuel via les cellules dites de Müller [1].
Un médicament de thérapie génique comme option de traitement pour les mutations du gène RPE65
L’idée de base est qu’en ajoutant une copie correcte par thérapie génique rétinienne, la fonction de la rétine peut être restaurée. Les vecteurs viraux adéno-associés recombinants (AAV) sont très bien adaptés pour effectuer le transfert de gènes, car ils permettent une expression prolongée et spécifique au type de cellule dans la rétine [9]. Voretigen Neparvovec est un vecteur de transfert de gènes qui utilise la capside d’un vecteur viral adéno-associé de sérotype 2 (AAV2) comme véhicule de transport de l’ADNc de la protéine rétinienne spécifique de l’épithélium pigmentaire humain de 65 kDa (hRPE65) dans la rétine [13].
| Prégénérique Neparvovec en cas de mutation biallélique du RPE65 L’efficacité du Luxturna® (Voretigen Neparvovec) a été étudiée chez 41 patients au total, tous atteints de dystrophie rétinienne héréditaire [13]. Dans l’étude principale, 11 adultes (36%) et 20 enfants de plus de 4 ans (64%) ont participé. L’âge moyen était de 15 ans. Afin de démontrer l’efficacité du Luxturna® dans l’étude principale, la vision fonctionnelle a été mesurée. Il s’agit de l’acuité visuelle, du champ visuel et de la perception et/ou de la capacité visuelle à faible luminosité. Après un an de traitement, les patients du bras de traitement Luxturna® étaient capables d’effectuer un parcours avec plus de précision, plus rapidement et dans des conditions d’éclairage plus faibles. L’amélioration de la vision fonctionnelle a été maintenue pendant une période d’au moins trois ans. Un suivi de 15 ans des 41 participants à l’étude est en cours. |
Luxturna® est un médicament de thérapie génique contenant le principe actif voreigen Neparvovec, autorisé en Suisse depuis février 2020 pour le traitement des adultes et des enfants atteints de perte de vision due à une dystrophie rétinienne héréditaire avec mutation RPE65 biallélique (encadré) [13]. Un traitement par Luxturna® suppose que la mutation du RPE65 a été confirmée par un test génétique et qu’il reste suffisamment de cellules fonctionnelles dans la rétine des patients [13]. Le Luxturna® est appliqué sous la rétine par microchirurgie [13,14]. En Suisse, un centre exclusif pour cette thérapie génique se trouve à l’Hôpital universitaire de Bâle (USB) [14]. Le professeur Hendrik Scholl est directeur et médecin-chef de la clinique ophtalmologique de l’USB, le professeur Christian Prünte y est médecin-chef clinique et s’est spécialisé entre autres dans les interventions microchirurgicales. En novembre 2022, l’USB a annoncé qu’une équipe de douze personnes, dirigée par les professeurs Scholl et Prünte, avait réalisé la première opération microchirurgicale de Suisse pour l’application de Luxturna®. Le patient de 51 ans atteint de dystrophie rétinienne héréditaire a vu son deuxième œil traité en mars 2022, après l’intervention de l’autre œil un an auparavant [14]. Les deux interventions se sont déroulées de manière optimale.
Messages Take-Home
- A ce jour, on connaît plus de 250 mutations qui peuvent être à l’origine d’une dystrophie rétinienne héréditaire. Dans 55 à 80 % des cas, la cause génétique peut être identifiée par des analyses génétiques moléculaires [4].
- Le défaut génétique sous-jacent définit la pathogenèse moléculaire, le tableau clinique et l’évolution de la maladie. Des variantes du gène RPE65 sont à l’origine de 0,6 à 6% de tous les cas de rétinite pigmentaire et de 3 à 16% des cas d’amaurose congénitale de Leber/dystrophie rétinienne à début précoce [2].
- En imagerie rétinienne, les mutations du gène RPE65 se caractérisent par une absence ou une diminution de l’autofluorescence au niveau du fond d’œil.
- Avec le Luxturna® (Voretigen Neparvovec), on dispose pour la première fois d’une thérapie génique pour le traitement de la dystrophie rétinienne héréditaire avec mutation RPE65 biallélique [13].
Littérature :
- Dias MF, et al. : Génétique moléculaire et thérapies émergentes pour la rétinite pigmentaire : recherche de base et perspectives cliniques. Progress in Retinal and Eye Research 2017 (63) : 107-131.
- Aoun M, et al. : Dystrophies rétiniennes héréditaires dues à des variants du RPE65 : Du diagnostic génétique au traitement. Kompass Ophthalmol 2021 ; 7 : 115-123.
- Nash BM, et al. : Dystrophies rétiniennes, applications génomiques dans le diagnostic et perspectives pour la thérapie. Translational Pediatrics 2015 ; 4 (2) : 139-163.
- Kohl S, Biskup S : Test de diagnostic génétique dans les dystrophies rétiniennes héréditaires. Klin Monbl Augenheilkd 2013 ; 230(3) : 243-246.
- Bolz HJ : Analyses diagnostiques des gènes de la dystrophie rétinienne : état actuel et perspectives. Klin Monbl Augenheilkd 2021 ; 238 : 261-266.
- Kellner U, et al : Diagnostic des dystrophies rétiniennes héréditaires. Place du diagnostic génétique moléculaire dans la perspective du patient. Ophthalmologie 2022 ; 119 : 820-826.
- Dockery A, et al : Next-generation sequencing applications for inherited retinal diseases. Int J Mol Sci 2021 ; 22 : 5684.
- Croix bleue et Bouclier bleu de Kansas City (BCBSKC) 2018. Thérapie génétique pour la dystrophie rétinienne héréditaire : Policy Number 2.04.144.
- Stieger K, Lorenz B : Thérapie génique pour les maladies dégénératives de la rétine, www.kaden-verlag.de/fileadmin/images/ZPA_direkt/CME_ZPA_12-09.pdf,(dernière consultation 04.04.2023)
- Birtel J, et al : Diagnostic des maladies héréditaires de la rétine Klin Monatsbl Augenheilkd 2021 ; 238 : 249-260.
- Robson AG, et al : Serial imaging and structure-function correlates of high-density rings of fundus autofluorescence in retinitis pigmentosa. Retina 2011 ; 31 : 1670-1679.
- Lima LH, et al : Constriction progressive de l’anneau hyperautofluorescent dans la rétinite pigmentaire. Am J Ophthalmol 2012 ; 153 : 718-727.
- Information sur les médicaments, www.swissmedicinfo.ch,(dernière consultation 04.04.2023)
- “Première utilisation de la thérapie génique contre la cécité en Suisse”, www.unispital-basel.ch/newscenter/zuweisenden-blog/15-07-2022,(dernière consultation 04.04.2023)
- Bjeloš M, et al. : RPE65 c.353G>A, p.(Arg118Lys) : Une nouvelle mutation ponctuelle associée à la rétinite pigmentaire et à l’atrophie maculaire. International Journal of Molecular Sciences. 2022 ; 23(18):10513. www.mdpi.com/1422-0067/23/18/10513,(dernière consultation 04.04.2023).
PRATIQUE DU MÉDECIN DE FAMILLE 2023 ; 18(4) : 46-47