Um doente adulto jovem com dores abdominais apresentava hepatomegalia e esplenomegalia associadas a trombocitopenia e anemia. A medula óssea foi então puncionada e biopsiada, seguindo-se a determinação da atividade enzimática da glucocerebrosidase leucocitária. O exame genético molecular acabou por confirmar o diagnóstico suspeito. Na população em geral, a prevalência da doença de Gaucher é de cerca de 1:100.000.
A doença de Gaucher é uma doença metabólica autossómica recessiva causada por uma mutação no gene da glucocerebrosidase e uma deficiência subsequente da enzima glucocerebrosidase. Como resultado, há uma acumulação de macrófagos carregados de lípidos (as chamadas células de Gaucher) no fígado e no baço, bem como na medula óssea, no sistema nervoso central (SNC) e nos pulmões [1]. Devido à sobrecarga crescente de glucocerebrosídeos, o fígado e o baço aumentam de tamanho e a medula óssea hematopoiética é deslocada [2]. A consequência é, por um lado, uma redução das plaquetas e dos eritrócitos e, por outro, uma destruição crescente da substância óssea.

Estudo de caso
Paciente do sexo feminino, 18 anos, foi admitida no hospital com queixa principal de dor no quadrante superior direito do abdómen, iniciada há quinze dias, de intensidade moderada e inicialmente com ligeira diminuição com anti-inflamatórios não esteróides (AINEs) orais [3]. Após algum tempo, o efeito analgésico dos AINEs diminuiu e, com o passar do tempo, parou completamente. Ao exame físico, a conjuntiva encontrava-se pálida e havia hepatomegalia dolorosa 5 cm abaixo do arco costal direito. As investigações complementares permitiram chegar às seguintes conclusões:
- Percussão: notava-se uma zona baça cerca de 8 cm abaixo do arco costal esquerdo, que se movia ao respirar. Este foi o fator decisivo para o internamento do doente no hospital.
- Exame ultrassonográfico do abdómen: O baço estava muito aumentado (20,1 × 8 cm), sem nódulos; o fígado estava mais de 6 cm abaixo do rebordo costal, sem nódulos; não havia gânglios linfáticos abdominais aumentados; pequena quantidade de líquido livre na cavidade peritoneal adjacente a alças intestinais de diâmetro normal; outros órgãos sem alterações.
- Aspirado de medula óssea: medula óssea hipercelular, com hiperplasia megacariopoiética e eritropoiética; integridade granulopoiética. Aumento de precursores de eosinófilos; aumento focal de linfócitos maduros e bem diferenciados; aumento de plasmócitos inferior a 10%; histiócitos abundantes com citoplasma claro semelhantes a células de Gaucher, sem sinais de hemofagocitose.
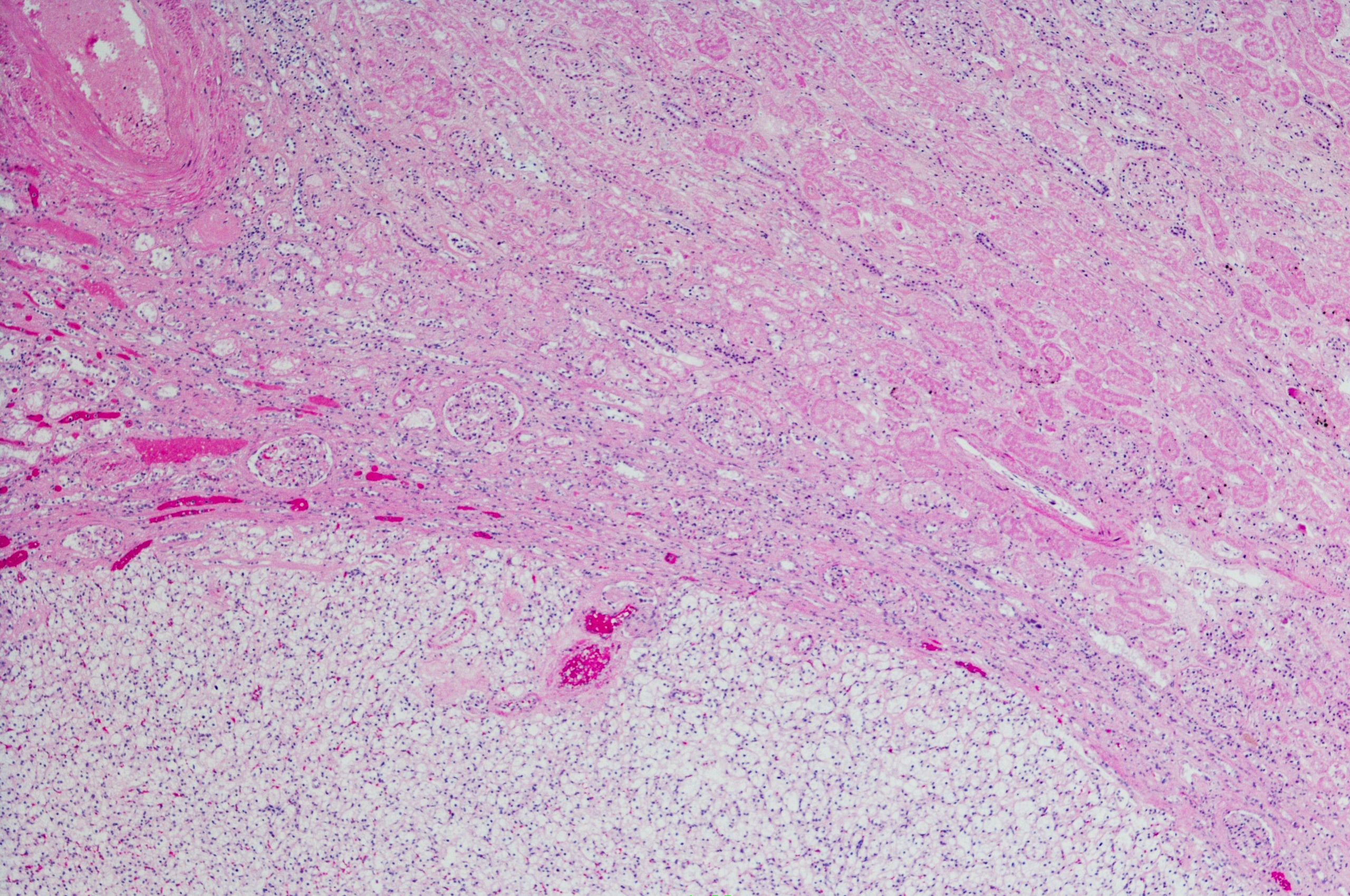
- Biópsia da medula óssea: Cilindros de medula óssea com mais de 6 espaços medulares com células histiocíticas mostrando citoplasma claro abundante em vacúolos mascarados por tecido hematopoiético remanescente. A aparência sugere uma doença de armazenamento lisossómico.
Após o resultado da biopsia, a atividade enzimática da glucocerebrosidase foi determinada em leucócitos periféricos. Este valor foi de 0,020 nmol/mg/h (valor normal: 0,12 nmol/mg/h). No exame genético molecular subsequente, foi finalmente detectada a dupla heterozigotia para as mutações L444P e N370S , pelo que o diagnóstico da doença de Gaucher pôde ser confirmado.
| Factos importantes sobre a M. Gaucher em resumo A doença de Gaucher pertence ao grupo das doenças de armazenamento lisossómico. Uma vez que os sintomas individuais podem também ter outras causas, o diagnóstico revela-se frequentemente difícil. O quadro clínico foi descrito pela primeira vez em 1882 por Philippe Charles Gaucher, que relatou um doente com esplenomegalia. Distinguem-se os seguintes três fenótipos clínicos da doença de Gaucher [2]: Tipo 1: forma não neuropática, Tipo 2: forma neuropática aguda, Tipo 3: forma neuropática crónica. Nos adultos, é sobretudo o tipo 1 ou 3 que se manifesta. Os sintomas típicos dos doentes com doença de Gaucher incluem dores nos ossos, diminuição do desempenho e tendência para sangrar. Além disso, a doença de Gaucher está associada a uma maior suscetibilidade a infecções [4]. O diagnóstico laboratorial revela tipicamente anemia e trombocitopenia. Os resultados imagiológicos são inovadores: a ecografia mostra um aumento do fígado e do baço e os raios X ou a ressonância magnética mostram alterações ósseas com destruição da estrutura da baelcae, bem como enfartes ósseos, que são visíveis pela primeira vez nos ossos tubulares longos das pernas. Raramente, a hipertensão pulmonar é encontrada devido ao armazenamento de glucocerebrosídeos nos macrófagos dos pulmões. Em caso de suspeita clínica de doença de Gaucher e de resultados laboratoriais e imagiológicos correspondentes, é aconselhável determinar a atividade enzimática da glucocerebrosidase num laboratório especializado. Atualmente, a terapia de substituição enzimática com glucocerebrosidase humana recombinante ou a terapia de redução do substrato estão disponíveis como abordagens de tratamento [2,5,6]. |
Literatura:
- Grabowski GA: Fenótipo, diagnóstico e tratamento da doença de Gaucher. Lancet 2008; 372: 1263-1271.
- Tran C, et al: Envolvimento pulmonar em pacientes adultos com erros inatos do metabolismo. Compass Pneumol 2018; 6 (1): 6-17.
- Valdés-Díaz K, et al: Doença de Gaucher. Apresentação de um caso clínico e revisão da literatura. Hematol Transfus Cell Ther 2022; 44(1): 104-107.
- Doença de Gaucher, www.uniklinik-duesseldorf.de/patienten-besucher/klinikeninstitutezentren/klinik-fuer-gastroenterologie-hepatologie-und-infektiologie/klinik/fuer-patienten/behandlungsschwerpunkte/stoffwechselkrankheiten/morbus-gaucher,(último acesso em 18.09.2023)
- Goitein O, et al: Lung involvement and enzyme replacement therapy in Gaucher’s disease (Envolvimento pulmonar e terapia de substituição enzimática na doença de Gaucher). QJM 2001; 94: 407-415.
- Shemesh E, et al: Terapia de substituição enzimática e redução de substrato para a doença de Gaucher. Cochrane Database Syst Rev 2015; 3: CD010324.
GP PRACTICE 2023; 18(9): 26