A un paciente adulto joven con dolor abdominal se le detectó hepatomegalia y esplenomegalia asociadas a trombocitopenia y anemia. A continuación se realizó una punción y biopsia de la médula ósea, seguida de la determinación de la actividad enzimática de la glucocerebrosidasa leucocitaria. El examen genético molecular confirmó finalmente el diagnóstico sospechado. En la población general, la prevalencia de la enfermedad de Gaucher es de aproximadamente 1:100.000.
La enfermedad de Gaucher es una enfermedad metabólica autosómica recesiva causada por una mutación en el gen de la glucocerebrosidasa y la consiguiente deficiencia de la enzima glucocerebrosidasa. Como resultado, se produce una acumulación de macrófagos cargados de lípidos (las llamadas células de Gaucher) en el hígado y el bazo, así como en la médula ósea, el sistema nervioso central (SNC) y los pulmones [1]. Debido a la sobrecarga creciente de glucocerebrósidos, el hígado y el bazo aumentan de tamaño y la médula ósea hematopoyética se desplaza [2]. La consecuencia es, por un lado, una reducción de las plaquetas y los eritrocitos y, por otro, una destrucción creciente de la sustancia ósea.

Estudio de caso
Una paciente de 18 años ingresó en el hospital con una queja principal de dolor abdominal en el cuadrante superior derecho que comenzó hace quince días con una intensidad moderada y que inicialmente se redujo ligeramente con antiinflamatorios no esteroideos (AINE) orales [3]. Al cabo de cierto tiempo, el efecto analgésico de los AINE disminuyó y, con el tiempo, cesó por completo. En la exploración física, la conjuntiva estaba pálida y había una hepatomegalia dolorosa 5 cm por debajo del arco costal derecho. Las investigaciones posteriores arrojaron los siguientes resultados:
- Percusión: Se observó una zona opaca a unos 8 cm por debajo del arco costal izquierdo, que se movía al respirar. Este fue el factor decisivo para el ingreso de la paciente en el hospital.
- Ecografía abdominal: Había un marcado agrandamiento del bazo (20,1 × 8 cm), sin nódulos; el hígado estaba a más de 6 cm por debajo del margen costal, sin nódulos; no había ganglios linfáticos abdominales agrandados; pequeña cantidad de líquido libre en la cavidad peritoneal adyacente a asas intestinales de diámetro normal; otros órganos sin cambios.
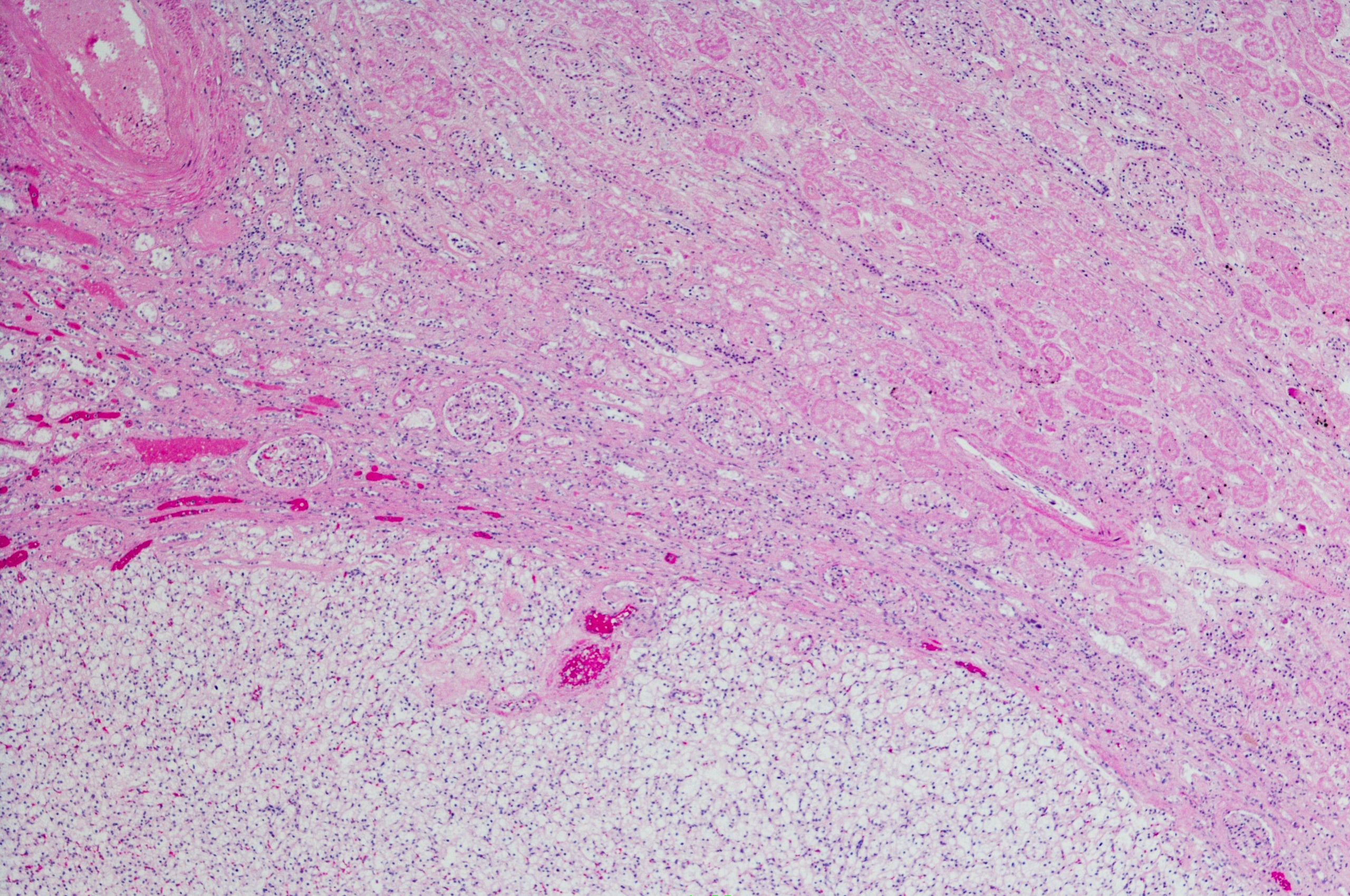
- Aspiración de médula ósea: Médula ósea hipercelular, con hiperplasia megacariopoyética y eritropoyética; integridad granulopoyética. Aumento de precursores de eosinófilos; aumento focal de linfocitos maduros bien diferenciados; aumento de células plasmáticas inferior al 10%; histiocitos abundantes con citoplasma claro que recuerdan a las células de Gaucher, sin signos de hemofagocitosis.
- Biopsia de médula ósea: Cilindros de médula ósea con más de 6 espacios medulares con células histiocíticas que muestran abundante citoplasma claro en vacuolas enmascaradas por el tejido hematopoyético restante. El aspecto sugiere una enfermedad por almacenamiento lisosómico.
Tras el resultado de la biopsia, se determinó la actividad enzimática de la glucocerebrosidasa en los leucocitos periféricos. Fue de 0,020 nmol/mg/h (valor normal: 0,12 nmol/mg/h). En el examen genético molecular posterior, se detectó finalmente una doble heterocigosidad para las mutaciones L444P y N370S , por lo que se pudo confirmar el diagnóstico de enfermedad de Gaucher.
| Datos importantes sobre M. Gaucher de un vistazo La enfermedad de Gaucher pertenece al grupo de enfermedades por almacenamiento lisosómico. Dado que los síntomas individuales también pueden tener otras causas, el diagnóstico a menudo resulta difícil. El cuadro clínico fue descrito por primera vez en 1882 por Philippe Charles Gaucher, que informó sobre un paciente con esplenomegalia. Se distinguen tres fenotipos clínicos de la enfermedad de Gaucher [2]: Tipo 1: forma no neuropática, Tipo 2: forma neuropática aguda, Tipo 3: forma neuropática crónica. En los adultos, son principalmente los tipos 1 o 3 los que se manifiestan. Los síntomas típicos de los pacientes con enfermedad de Gaucher incluyen dolor óseo, disminución del rendimiento y tendencia a las hemorragias. Además, la enfermedad de Gaucher se asocia a una mayor susceptibilidad a las infecciones [4]. El diagnóstico de laboratorio suele mostrar anemia y trombocitopenia. Los hallazgos por imagen son revolucionarios: la ecografía muestra un agrandamiento del hígado y el bazo, y las radiografías o resonancias magnéticas muestran cambios óseos con destrucción de la estructura baelcae, así como infartos óseos, que son visibles por primera vez en los huesos tubulares largos de las piernas. En raras ocasiones, la hipertensión pulmonar se debe al almacenamiento de glucocerebrósidos en los macrófagos de los pulmones. En caso de sospecha clínica de enfermedad de Gaucher y los correspondientes hallazgos de laboratorio e imagen, es aconsejable determinar la actividad enzimática de la glucocerebrosidasa en un laboratorio especializado. Hoy en día, la terapia de sustitución enzimática con glucocerebrosidasa humana recombinante o la terapia de reducción de sustrato están disponibles como enfoques de tratamiento [2,5,6]. |
Literatura:
- Grabowski GA: Fenotipo, diagnóstico y tratamiento de la enfermedad de Gaucher. Lancet 2008; 372: 1263-1271.
- Tran C, et al: Afectación pulmonar en pacientes adultos con errores innatos del metabolismo. Compass Pneumol 2018; 6 (1): 6-17.
- Valdés-Díaz K, et al: Enfermedad de Gaucher. Presentación de un caso clínico y revisión de la literatura. Hematol Transfus Cell Ther 2022; 44(1): 104-107.
- Enfermedad de Gaucher, www.uniklinik-duesseldorf.de/patienten-besucher/klinikeninstitutezentren/klinik-fuer-gastroenterologie-hepatologie-und-infektiologie/klinik/fuer-patienten/behandlungsschwerpunkte/stoffwechselkrankheiten/morbus-gaucher,(último acceso 18.09.2023)
- Goitein O, et al: Afectación pulmonar y terapia de sustitución enzimática en la enfermedad de Gaucher. QJM 2001; 94: 407-415.
- Shemesh E, et al: Terapia de sustitución enzimática y reducción de sustrato para la enfermedad de Gaucher. Cochrane Database Syst Rev 2015; 3: CD010324.
PRÁCTICA GP 2023; 18(9): 26