As ictioses constituem um grupo heterogéneo de doenças cutâneas hereditárias raras cuja classificação pode ser difícil. Por conseguinte, o diagnóstico rápido e o início da terapêutica constituem um desafio na prática clínica quotidiana. Os peritos da Clínica de Doenças da Pele do Hospital Universitário de Münster, especializados em genodermatoses, resumiram as características dermatopatológicas dos diferentes subtipos de ictiose num artigo de síntese. A revisão foi publicada na revista Dermatopathology.
Todas as ictioses (do grego “ichthys”=peixe) têm em comum defeitos em componentes-chave da barreira epidérmica, resultando numa perturbação da cornificação da pele. Esta manifesta-se por uma descamação visível e/ou hiperqueratose da pele, com expressão clínica e etiologia heterogéneas. Podem também ocorrer bolhas e componentes inflamatórios. A evolução depende do tipo de ictiose e das possíveis complicações (por exemplo, prurido, infecções cutâneas).
Ictiose vulgaris
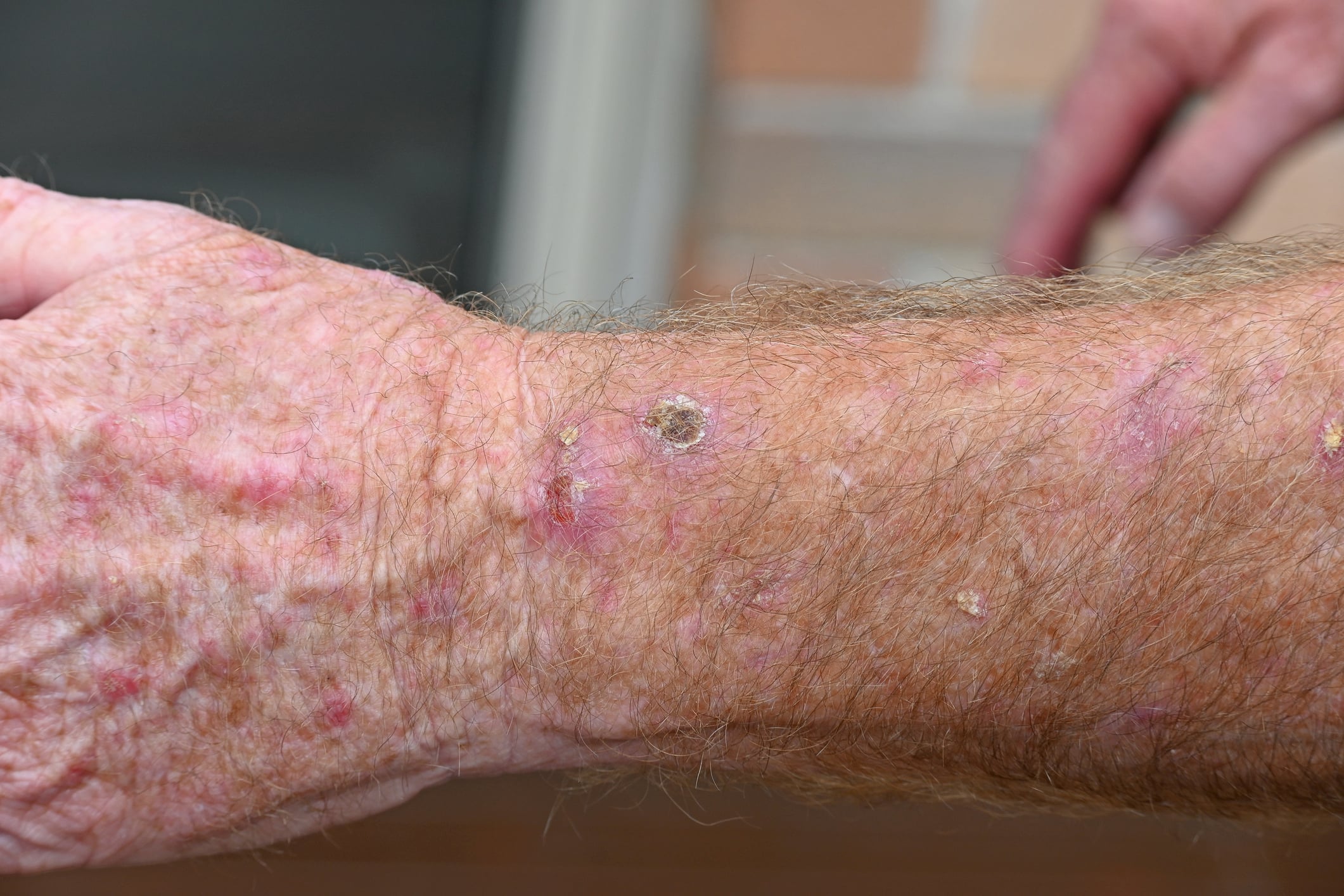
A ictiose vulgar autossómica de herança semidominante é a forma mais comum de ictiose [2]. Desenvolve-se normalmente durante o primeiro ano de vida e manifesta-se por pele seca ou escamas finas cinzentas claras (Fig. 1) e hiperlinearidade palmoplantar.

Histologia: Uma característica histológica é um estrato granuloso severamente reduzido, muitas vezes completamente ausente. O estrato córneo apresenta uma ligeira orto-hiperqueratose compacta (Fig. 2) . A hiperqueratose dos folículos pilosos e a acrosiringe são também frequentemente encontradas. A epiderme pode ser ligeiramente alargada, mas também atrófica. A derme apresenta esporadicamente infiltrados linfocíticos perivasculares ligeiros. Imunohistoquimicamente, pode ser quantificada uma deficiência de filagrina, que se correlaciona com o número de mutações no gene da filagrina e, por conseguinte, com a gravidade da ictiose. Ultra-estruturalmente, existe um defeito nos grânulos de querato-hialina, que aparecem reduzidos em tamanho e friáveis.

Ictioses congénitas autossómicas recessivas
As ictioses congénitas autossómicas recessivas (ARCI) representam um grupo geneticamente heterogéneo de ictioses não sindromáticas com uma gravidade muito variável. As formas ARCI são causadas por diferentes mutações e incluem a ictiose lamelar, a eritrodermia ictiosiforme congénita e o subtipo mais grave, mas raro, de ictiose arlequim [3]. Os recém-nascidos podem nascer com um estrato córneo apertado e brilhante, que está associado a ectrópio, eclábio, perda de fluidos e desregulação térmica, levando a complicações potencialmente fatais. No entanto, a característica clínica de uma membrana de colódio também é observada noutras ictioses [4]. Mais tarde, a membrana de colódio é substituída por escamas castanho-escuras, aderentes e semelhantes a placas(ictiose lamelar clássica; Figura 3) ou por uma escamação fina e esbranquiçada na pele avermelhada (eritrodermia ictiosiforme congénita não bolhosa).

Histologia: Há uma orto-hiperqueratose compacta, um estrato granuloso ligeiramente alargado, acantose e papilomatose da epiderme. No corpo papilar, os vasos aparecem dilatados e em espiral, e os infiltrados linfocíticos são esparsos ou reduzidos (Fig. 4) [5].

Ictiose queratinopática
A ictiose epidermolítica deve-se a mutações na queratina 1 ou na queratina 10 e é, por isso, classificada como ictiose queratinopática [4]. Nos recém-nascidos, ocorre eritrodermia com bolhas por vezes pronunciadas, e mais tarde desenvolvem-se hiperqueratoses (pontiagudas), preferencialmente nas extremidades (Fig. 5). Os doentes com mutações da queratina 1 também têm queratose palmo-plantar, o que não ocorre em doentes com mutações da queratina 10, uma vez que esta queratina não é expressa nestes doentes [6].

Histologia: Há uma orto-hiperqueratose maciça e acantose da epiderme. Os queratinócitos suprabasais apresentam vacuolização e grânulos hipereosinofílicos distintos. No estrato granuloso, os grânulos de querato-hialina são grosseiros e irregulares. Os limites entre os queratinócitos são mal demarcados e aparecem fissuras e bolhas. Podem ocorrer infiltrados linfocíticos mais pequenos na derme [7].
Ao microscópio electrónico, os grânulos hipereosinofílicos correspondem a aglomerados do esqueleto de queratina. O colapso das queratinas mutantes provoca o aspecto vacuolar do citoplasma e conduz à instabilidade mecânica.
Eritroqueratodermia
A eritroqueratodermia caracteriza-se por queratoses eritematosas localizadas no corpo e está actualmente classificada no grupo das ictioses [4]. Baseiam-se em mutações dos genes da conexina 31 (GJB3) e da conexina 30.3 (GJB4). As conexinas são proteínas transmembranares nas junções comunicantes que são essenciais para a comunicação intercelular e, por conseguinte, para a diferenciação epidérmica [8]. A eritroqueratodermia variável (síndrome de Mendes da Costa), que é herdada de forma autossómica dominante, manifesta-se primeiro com eritema errante de ângulo fino e mais tarde com queratoses persistentes. A expressão varia entre intra e interfamiliar e, por vezes, apenas se encontram queratoses circunscritas em áreas expostas à pressão na planta do pé.
Histologia: Histologicamente, a epiderme mostra acantose e uma superfície ondulada com hiperqueratose, paraqueratose focal, queratinócitos disqueratóticos e estrato granuloso preservado. Pode estar presente um infiltrado linfocítico perivascular superficial (Fig. 6). Em geral, as alterações histológicas mencionadas são muito variáveis, o que complica o diagnóstico. A deficiência da conexina afectada pode ser facilmente visualizada em imunohistoquímica; ao mesmo tempo, a expressão compensatória da conexina 43 está aumentada.

Síndrome KID e síndrome HID
A síndrome da surdez por queratite ictiose (síndrome KID) e a síndrome da surdez por ictiose tipo hystrix (síndrome HID) são formas diferentes de uma ictiose hereditária autossómica dominante causada por uma mutação na conexina-26 [9]. Uma vez que esta conexina desempenha funções importantes no ouvido interno, também se regista perda auditiva neurossensorial. Os doentes com a síndrome KID desenvolvem placas hiperqueratóticas verrucosas nitidamente circunscritas na face e nas extremidades; na síndrome HID, predomina a ictiose generalizada do tipo hystrix. No contexto desta ictiose sindrómica, surgem queratites, alopécia, distrofia ungueal, anomalias dentárias ou hipoidrose, bem como um risco acrescido de infecção e de carcinoma.
Histologia: A epiderme é acantótica e tem um aspecto parcialmente verruciforme. O estrato córneo hiperqueratótico contém paraqueratoses com grandes restos nucleares redondos e, ocasionalmente, células-sombra com núcleos vacuolados (Fig. 7) . Os queratinócitos disqueratóticos com halo perinuclear (“olho de pássaro”) aparecem como critério dominante. O estrato granuloso pode estar ausente ou fortemente pronunciado. Nalguns casos, ocorrem infiltrados linfocíticos densos subepidérmicos. As aberturas dos folículos pilosos e das glândulas sudoríparas são altamente queratinizadas. As glândulas sudoríparas podem ser reduzidas em tamanho e atróficas. Os carcinomas de células escamosas altamente diferenciados também podem ocorrer numa idade jovem [7].

Síndrome de Netherton
Na síndrome de Netherton, autossómica recessiva, existe uma mutação do gene SPINK5, que codifica o LEKTI (“lymphoepithelial Kazal-type related inhibitor”), um importante inibidor da serina protease da epiderme e do timo [10]. Os doentes nascem com uma eritrodermia pronunciada, que mais tarde se transforma frequentemente em eritema anular com linhas e áreas escamosas típicas em forma de anel (“ictiose linear circunflexa”) (Fig. 8). Na evolução tardia, pode ocorrer cabelo quebradiço (“cabelo de bambu”, tricorrexis invaginata), alergias de tipo 1, aumento dos níveis de IgE e hipereosinofilia, bem como imunodeficiência e enteropatia, que podem ser acompanhadas por um enorme atraso no desenvolvimento, especialmente no primeiro ano de vida. Os distúrbios electrolíticos e a sépsis representam um risco fatal para os bebés.

Histologia: Há uma hiperplasia psoriasiforme com um estrato córneo moderadamente alargado com paraqueratose focal e acumulações de neutrófilos. O estrato granuloso está ausente ou severamente reduzido. A derme papilar está alargada e contém vasos dilatados e infiltrados inflamatórios com linfócitos, neutrófilos e granulócitos eosinofílicos (Fig. 9) . Por vezes, no entanto, ocorrem alterações histológicas, como na dermatite atópica. Imunohistoquimicamente, a coloração para LEKTI está tipicamente ausente na epiderme e nos folículos pilosos [11].

“Síndrome da pele descamada
A síndrome da pele descamada é uma forma inflamatória rara de ictiose. Desde o nascimento, observa-se um eritema generalizado com descolamento superficial da pele, que persiste com variações sazonais ao longo da vida (Fig. 10). Além disso, pode ocorrer um descolamento episódico das placas ungueais (onicomadese). O estado dos pêlos é discreto, à excepção de uma depilação ligeira temporária dos pêlos finos [12]. Existem mutações no gene CDSN, que codifica a proteína corneodesmosina. A corneodesmosina é uma importante proteína de adesão expressa nas secções extracelulares dos desmossomas no estrato córneo da epiderme e na bainha interna da raiz do cabelo dos folículos pilosos. Ultra-estruturalmente, verifica-se o descolamento de corneócitos intactos do estrato granuloso (clivagem extracelular) [12,13]. A ruptura concomitante da barreira leva a inflamação com prurido maciço, urticária, angioedema, alergias alimentares e asma com níveis aumentados de IgE e eosinofilia no sangue.

Histologia: A epiderme é hiperplásica e tem cristas de rete pronunciadas. Existe uma hiperqueratose ligeira com paraqueratose focal e um estrato granuloso mais fino. Algumas biopsias mostram um descolamento focal do estrato córneo e, nalguns casos, o estrato córneo está completamente ausente. No entanto, estas alterações nem sempre podem ser vistas numa secção de parafina. Existem infiltrados linfocíticos superficiais e perivasculares com neutrófilos isolados também encontrados no estrato córneo. O corpo papilar é alongado e edematoso, os vasos não estão dilatados [5]. Imunohistoquimicamente, a coloração para a corneodesmosina [12] está ausente no estrato córneo.
Síndrome da criança
A síndrome CHILD (“Congenital Hemidysplasia with Ichthyosiform nevus and Limb Defects”) é uma genodermatose dominante ligada ao X com lesões cutâneas inflamatórias e escamosas unilaterais e malformações ipsilaterais dos órgãos internos e das extremidades. que é normalmente fatal para a descendência masculina. Caracteriza-se por lesões cutâneas inflamatórias unilaterais, frequentemente cerosas e amarelas, que ocorrem principalmente nas grandes flexuras e na região perianogenital [7,14]. Os sintomas extracutâneos variam desde hipoplasia discreta dos membros até deformidades graves. Os casos monossintomáticos são frequentemente diagnosticados erradamente como psoríase ou ILVEN (nevo epidérmico verrucoso linear inflamatório) [23]. A doença é causada por mutações sem sentido ou de sentido errado no chamado gene NSDHL [16,17].
Histologia: Há uma epiderme hiperplásica com cristas de rete alongadas e orto-hiperqueratose pronunciada com paraqueratose focal. O estrato granuloso pode ser pronunciado em algumas áreas ou pode estar ausente. Um infiltrado linfocítico perivascular e macrófagos xantomatosos são observados no corpo papilar [5].
Síndrome SAM
A síndrome SAM (“Severe dermatitis, multiple allergies, metabolic wasting syndrome”) foi descrita em 2013 por Samuelov et al. identificada como uma genodermatose grave, potencialmente fatal, causada por uma mutação na desmogleína-1 (DSG1) ou na desmoplaquina [18,19]. A doença é herdada de uma forma autossómica recessiva. Clinicamente, os recém-nascidos apresentam eritrodermia ictiosiforme, semelhante à ictiose lamelar autossómica recessiva, à síndrome de Netherton ou à síndrome da “pele a descascar”. Outros sintomas incluem prurido, hipotricose, alergias alimentares, disfagia, crescimento reduzido e infecções cutâneas e respiratórias recorrentes. Existem vários graus de formação pustular, queratoses palmo-plantares, onicodistrofia, anomalias dentárias, anomalias cardíacas e esofagite eosinofílica. Existe uma grande variabilidade inter e intra-familiar [20]. A inflamação que a acompanha pode ser explicada pela actividade pró-inflamatória dos queratinócitos associada a uma função de barreira prejudicada e a um bloqueio desregulado das vias de transdução de sinal [15,21,22]. Tal como na síndrome de Netherton, as terapias anti-inflamatórias com produtos biológicos melhoram o quadro clínico.

Histologia: Há uma dermatite linfocítica superficial com epiderme hiperplásica, paraqueratose e granulócitos neutrofílicos que lembram a dermatite psoriasiforme. No entanto, os espaços intercelulares dilatados na epiderme sem formação de bolhas são típicos (Fig. 11) . Estão ausentes o edema intracelular da epiderme (dermatite esponjosa), o arredondamento dos queratinócitos e a picnose dos núcleos (pênfigo), a hipereosinofilia do citoplasma (doença de Darier ou Hailey-Hailey), as alterações nucleares viropáticas (herpes).
Literatura:
- Metze D, Traupe H, Süssmuth K: Ichthyoses – A Clinical and Pathological Spectrum from Heterogeneous Cornification Disorders to Inflammation. Dermatopathology. 2021; 8(2): 107–123. www.mdpi.com/2296-3529/8/2/17, (letzter Abruf 06.04.2023)
- Majmundar VD, Baxi K: Hereditary and Acquired Ichthyosis Vulgaris. In Treasure Island (FL).; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Hotz A, et al.: Meta-Analysis of Mutations in ALOX12B or ALOXE3 Identified in a Large Cohort of 224 Patients. Genes 2021; 12: 80
- Oji V, et al.: JAAD 2010; 63: 607–641.
- Metze D: Disorders of Keratinization. In McKee’s Pathology of the Skin, 5th ed.; 2-Volume-Set; Calonje E, et al. (Eds.); Elsevier: Amsterdam, The Netherlands, 2019; Volume 1, Chapter 3; pp. 53–117.
- Rothnagel JA, et al.: Science 1992; 257: 1128–1130.
- Oji V, Metze D, Traupe H: Inherited disorders of cornification. In Rook’s Textbook of Dermatology, 9th ed.; Burns T, (Eds.); Wiley-Blackwell: Hoboken, NJ, USA, 2016; Volume 2, Part 6; Chapter 65; pp. 1–75.
- Avshalumova L, Fabrikant J, Koriakos A: J Dermatol 2014; 53: 192–205.
- van Geel M, et al.: BJD 2002; 146: 938–942.
- Chavanas S, et al.: Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet 2000; 25: 141–142.
- Leclerc-Mercier S, et al.: Am J Dermatopathol 2016; 38: 83–91.
- Oji V, et al.: Am J Hum Genet 2010; 87: 274–281.
- Süßmuth K, et al.: JDDG 2020; 18: 225–243.
- Ramphul K, Kota V, Mejias SG: Child Syndrome. In Treasure Island (FL); StatPearls Publishing: Treasure Island, FL, USA, 2021
- Polivka L, et al.: JACI 2018; 142: 702–706.e7.
- Bergqvist C, et al.: Am J Med Genet A 2018; 176: 733–738.
- König A, et al.: Am J Med Genet 2000; 90: 339–346.
- Samuelov L, et al.: Nat Genet 2013; 45: 1244–1248.
- McAleer MA, et al.: JACI 2015; 136: 1268–1276.
- Taiber S, et al.: Exp Dermatol 2018; 27: 787–790.
- Hammers CM, Stanley JR: J Clin Investig 2013; 123: 1419–1422.
- Ishida-Yamamoto A, Igawa S: J Dermatol Sci 2014; 74: 99–105.
- Happle R, Koch H, Lenz W: Eur J Pediatr 1980; 134: 27–33.
DERMATOLOGIE PRAXIS 2023; 33(2): 42–44