A síndrome de Clouston é uma doença hereditária autossómica dominante caracterizada pelos principais sintomas de hiperqueratose, distrofia das unhas e alopecia. A síndrome é causada por mutações no gene GJB6, que codifica a junção da proteína connexin-30.
Esta doença rara foi descrita pela primeira vez em 1929 pelo médico canadiano H.R. Clouston. A prevalência exacta não é conhecida, provavelmente a síndrome nem sempre é diagnosticada. O sintoma mais regular são as alterações das unhas, que podem estar presentes no nascimento ou no início da vida neonatal. Normalmente, os pregos são espessados, crescem lentamente, e são quebradiços, frequentemente hiperconvexos, descoloridos e listrados. Os sintomas adicionais são microoníquia, onicólise e paroníquia recorrente com perda das unhas. Outro sintoma característico é a alopecia total a parcial, muitas vezes progressiva, que já está presente à nascença ou aparece na infância/criança. O restante cabelo principal cresce lentamente e é esparso, fino e quebradiço. Sobrancelhas, pestanas e pêlos do corpo também são frequentemente esparsos. A hiperqueratose palmo-plantar não ocorre regularmente. Se presente, geralmente começa na infância e tende a piorar com o avanço da idade. Alguns pacientes também desenvolvem hiperqueratose e hiperpigmentação sobre as articulações e proeminências ósseas.
Casuística: Exemplo da vida hospitalar quotidiana
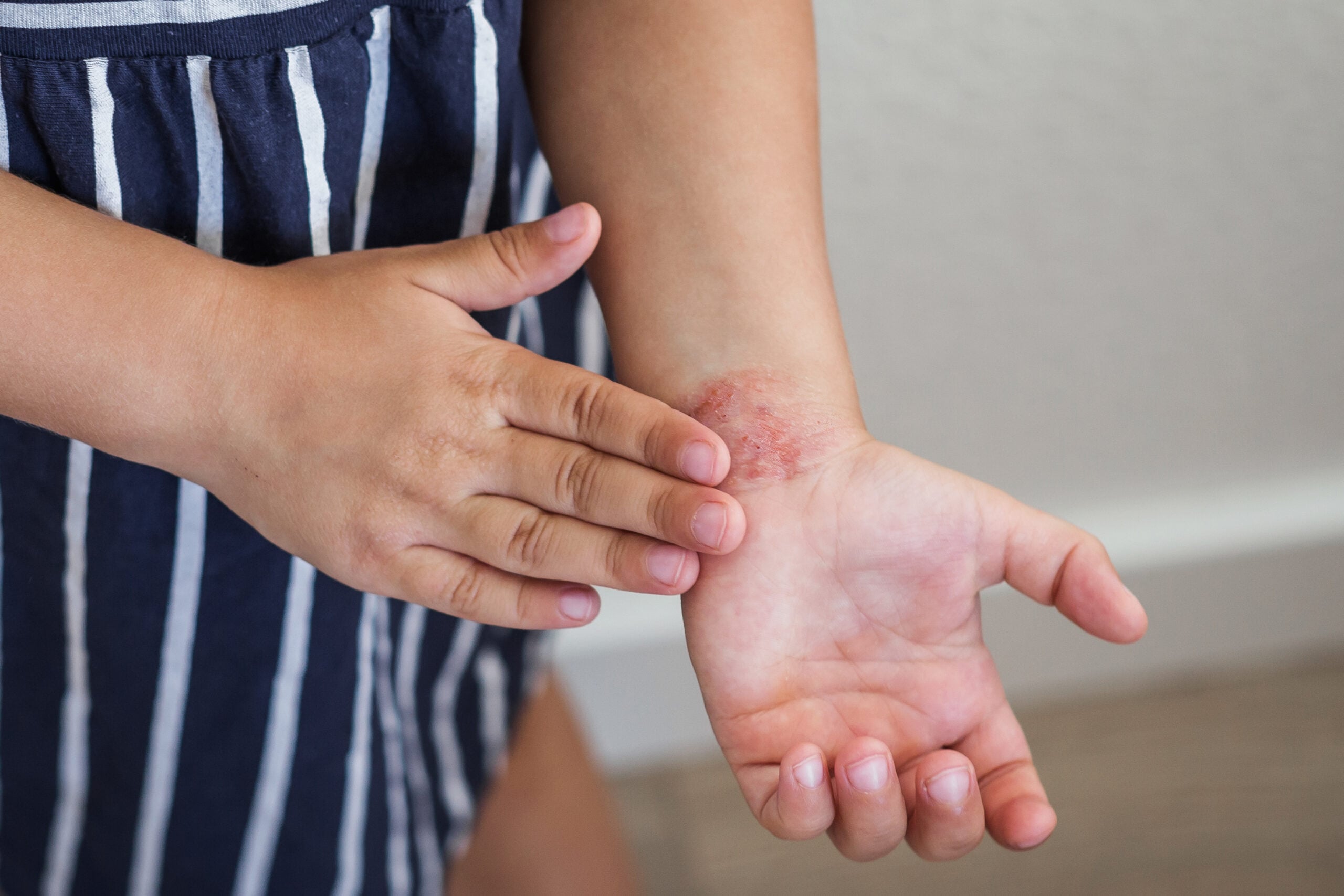
Uma equipa de investigação do departamento de dermatologia do Hospital Universitário de Lausanne descreveu o seguinte exemplo de uma família com raízes canadianas: a rapariga de 4 anos tem cabelo frágil, quebradiço e hipotricose, bem como unhas hiperqueratósicas e palmas e solas ligeiramente avermelhadas e espessadas. No pai, as lesões palmoplantar estendem-se até aos limites dos pés e das mãos, o tendão de Aquiles e os tornozelos. Se se formarem fissuras, a dor ocorre ao caminhar. Problemas semelhantes ocorreram ao longo de, pelo menos, quatro gerações. Alguns dos membros da família eram carecas, enquanto outros tinham mais queratodermia palmo-plantar e anomalias nas unhas, e outros apresentavam quase nenhum sintoma. Os testes genéticos moleculares revelaram uma mutação heterozigótica no gene GJB6. A biopsia de pele mostrou hiperqueratose mas quase nenhuma alteração inflamatória, o que foi confirmado pela ausência de uma assinatura inflamatória das análises transcriptómicas.

Opções de tratamento
Até agora, só existem opções de tratamento sintomático para a síndrome de Clouston. Estes incluem queratolíticos tópicos, retinóides orais, e dispositivos ortopédicos para reduzir a pressão. Num modelo animal utilizando ratos adultos Cx30A88V/A88V, um anticorpo contra a connexina 30 levou a uma redução na actividade de hemicanal de junção de fendas. De acordo com Morren et al. esta pode ser uma nova abordagem de tratamento. As connexinas são uma família de proteínas transmembranas que formam junções entre células e permitem a troca directa de moléculas até um tamanho de cerca de 1 kDa entre células vizinhas.
Fontes:
- Orphanet, www.orpha.net,(último acesso 09.02.2023)
- Morren MA, et al: Síndrome de Clouston: uma forma rara de displasia ectodérmica. Poster 15, volume abstracto, Reunião Anual da SGDV, 9-11.11.2022.
DERMATOLOGIE PRAXIS 2023; 33(1): 37 (publicado 14.2.23, antes da impressão).