El síndrome de Clouston es un trastorno hereditario autosómico dominante caracterizado por los síntomas principales de hiperqueratosis, distrofia ungueal y alopecia. El síndrome está causado por mutaciones en el gen GJB6, que codifica la proteína de la unión en hendidura conexina-30.
Esta rara enfermedad fue descrita por primera vez en 1929 por el médico canadiense H.R. Clouston. No se conoce la prevalencia exacta, probablemente el síndrome no siempre se diagnostica. El síntoma más habitual son los cambios en las uñas, que pueden presentarse al nacer o en la vida neonatal temprana. Normalmente, las uñas están engrosadas, crecen lentamente y son quebradizas, a menudo hiperconvexas, descoloridas y rayadas. Otros síntomas son la microoniquia, la onicólisis y la paroniquia recurrente con pérdida de las uñas. Otro síntoma característico es la alopecia total o parcial, a menudo progresiva, que ya está presente al nacer o aparece en la infancia/niñez. El resto del vello principal crece lentamente y es escaso, fino y quebradizo. Las cejas, las pestañas y el vello corporal también suelen ser escasos. La hiperqueratosis palmoplantar no se produce con regularidad. Si está presente, suele comenzar en la infancia y tiende a empeorar con el avance de la edad. Algunos pacientes también desarrollan hiperqueratosis e hiperpigmentación sobre las articulaciones y las prominencias óseas.
Casuística: Ejemplo de la vida hospitalaria cotidiana
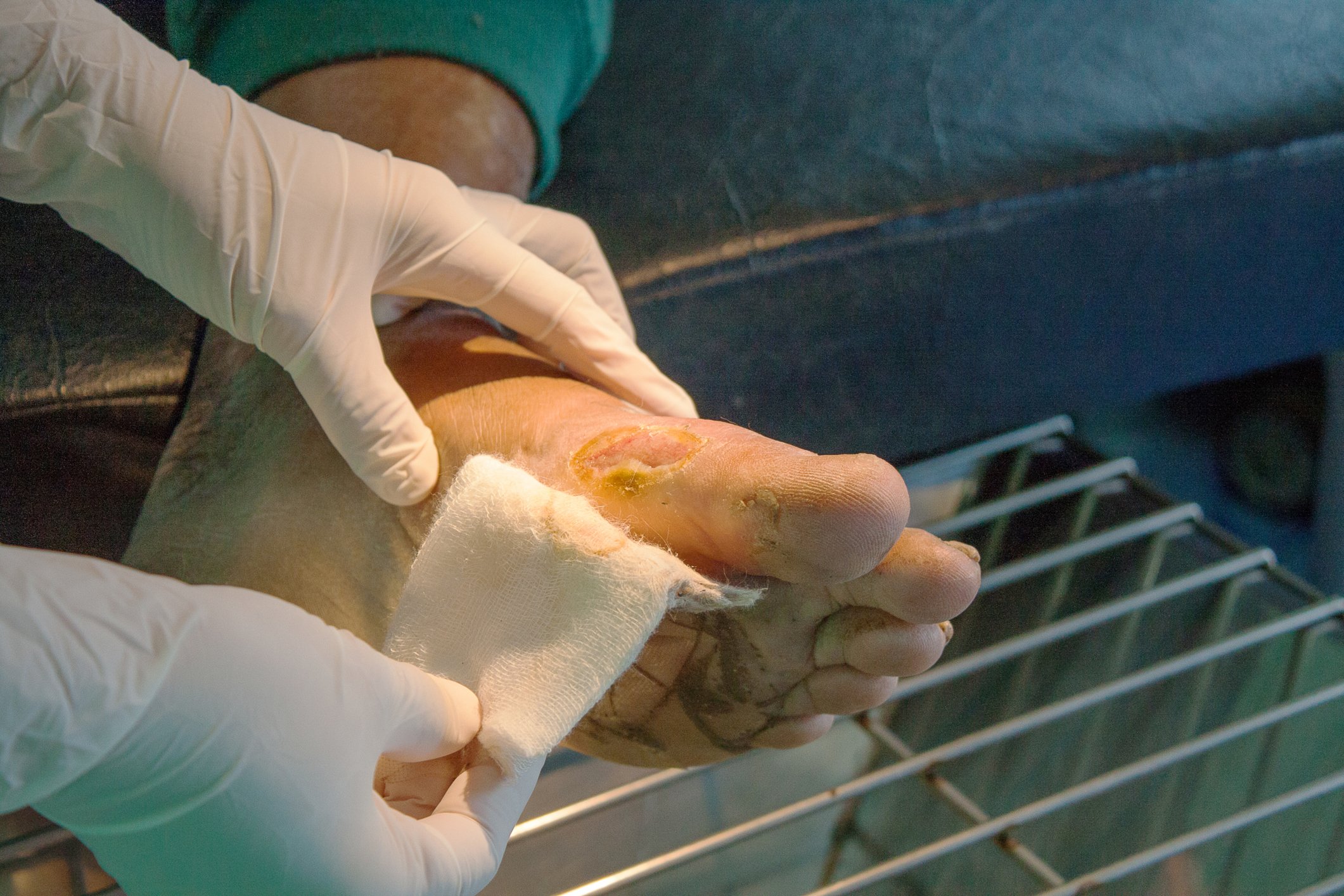
Un equipo de investigación del departamento de dermatología del Hospital Universitario de Lausana describió el siguiente ejemplo de paciente de una familia con raíces canadienses: la niña de 4 años tiene el pelo quebradizo y quebradizo e hipotricosis, así como uñas hiperqueratósicas y palmas y plantas ligeramente enrojecidas y engrosadas. En el padre, las lesiones palmoplantares se extienden a los bordes de los pies y las manos, el tendón de Aquiles y los tobillos. Si se forman grietas, se produce dolor al caminar. Se han producido problemas similares durante al menos cuatro generaciones. Algunos de los miembros de la familia eran calvos, mientras que otros presentaban más queratodermia palmoplantar y anomalías en las uñas, y otros apenas mostraban síntomas. Las pruebas genéticas moleculares revelaron una mutación heterocigota en el gen GJB6. La biopsia cutánea mostró hiperqueratosis pero apenas cambios inflamatorios, lo que se confirmó por la ausencia de una firma inflamatoria en los análisis transcriptómicos.

Opciones de tratamiento
Hasta ahora, sólo existen opciones de tratamiento sintomático para el síndrome de Clouston. Entre ellos se incluyen los queratolíticos tópicos, los retinoides orales y los dispositivos ortopédicos para reducir la presión. En un modelo animal con ratones adultos Cx30A88V/A88V, un anticuerpo contra la conexina 30 provocó una reducción de la actividad de los hemicanales de la unión gap. Según Morren et al. este puede ser un nuevo enfoque de tratamiento. Las conexinas son una familia de proteínas transmembrana que forman uniones gap en las células y permiten el intercambio directo de moléculas de hasta un tamaño de aproximadamente 1 kDa entre células vecinas.
Fuentes:
- Orphanet, www.orpha.net,(último acceso 09.02.2023)
- Morren MA, et al: Síndrome de Clouston: una forma rara de displasia ectodérmica. Póster 15, volumen de resúmenes, Reunión anual de la SGDV, 9-11.11.2022.
DERMATOLOGIE PRAXIS 2023; 33(1): 37 (publicado el 14.2.23, antes de impresión).