La atrofia muscular espinal es una enfermedad genética rara caracterizada por la pérdida de neuronas motoras en la médula espinal y la parte inferior del tronco encefálico. Mientras tanto, los expertos coinciden en que la gestión de la terapia debe orientarse a las necesidades individuales y los objetivos subjetivos de la persona afectada, especialmente a medida que aumenta la edad. Porque éstas tienen una gran influencia en la calidad de vida.
Uno de cada 10.000 recién nacidos tiene un defecto genético que altera la transmisión del impulso de las neuronas motoras. La atrofia muscular espinal (AME) es una enfermedad muscular rara, predominantemente autosómica recesiva, en la que degeneran las células motoras del asta anterior y los núcleos motores de los nervios craneales. Es la enfermedad hereditaria más común que provoca la muerte en la infancia y suele diagnosticarse a una edad muy temprana, aunque no exclusivamente [1,2]. La AME también puede manifestarse por primera vez en la edad adulta [3]. Esta heterogeneidad requiere, por tanto, una terapia individual que tenga en cuenta la actividad de la enfermedad y las necesidades del paciente.
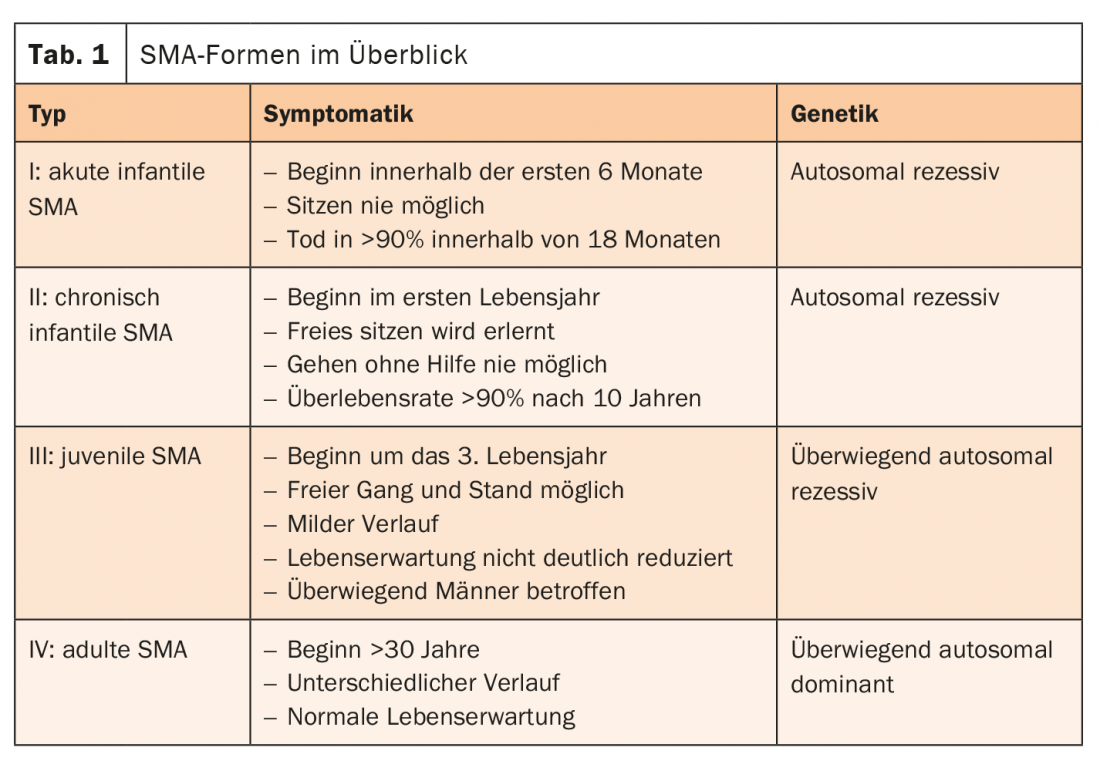
La causa de la AME suele ser un defecto en el gen SMN1. Junto con el gen SMN2, forma la proteína “Supervivencia de la neurona motora” (SMN). Ésta desempeña un papel central en la transmisión de los impulsos de las células nerviosas a los músculos. Si el gen SMN1 falla, la importante proteína sólo puede ser producida por el gen SMN2 restante. Por lo tanto, cuantas más copias de SMN2 haya, más tardía será la aparición de la AME y más favorable su evolución. Los pacientes con AME de aparición tardía (tipo II-IV) suelen tener una esperanza de vida normal. Las formas de AME se diferencian según el patrón de distribución, el inicio de la enfermedad, la gravedad y el patrón hereditario (Tab. 1) [4].
Cribar a los recién nacidos si es posible
Un diagnóstico definitivo sobre la AME sólo puede hacerse mediante pruebas genéticas. Sin embargo, dado que la AME tipo 1, si no se trata, conduce a la muerte o requiere ventilación mecánica permanente en el 90% de los casos para cuando el paciente alcanza los dos años de edad, está indicada la detección precoz de la enfermedad con el inicio más rápido posible del tratamiento. La gestión terapéutica es compleja e incluye cuidados agudos, así como medidas relativas a la rehabilitación, ortopedia, asistencia respiratoria, fisioterapia e intervenciones farmacológicas.
Éxitos terapéuticos también con adolescentes y adultos
El primer fármaco que no sólo actúa sintomáticamente sino que también aborda las causas de la enfermedad corrigiendo el defecto genético subyacente fue aprobado en 2017 con el nusinersen. El oligonucleótido antisentido (ASO) es un modulador específico del splicing que deja el genoma tal cual y potencia la función natural de la proteína SMN2. Esto permite que se formen mayores cantidades de proteína SMN completa y funcional. Ahora, se han presentado los primeros datos de un estudio de cohortes multicéntrico. El objetivo principal de este estudio observacional no intervencionista es investigar los objetivos y las expectativas de tratamiento de los pacientes adultos con 5q-SMA, así como la satisfacción subjetiva con el tratamiento de los pacientes [5].
Los resultados preliminares muestran que los objetivos individuales del tratamiento son de especial importancia. Dentro de los distintos tipos de 5q-SMA, los objetivos de la terapia varían significativamente. Así, la preservación de la función del brazo es uno de los objetivos más comunes del tratamiento, con predominio en los pacientes de tipo 1 y 2. En los pacientes con AME de tipo 3, se suele dar prioridad a la conservación y mejora de la función de las piernas. La terapia ASO parece adecuada para ello. Los resultados de otros estudios también muestran que varias capacidades motoras pueden estabilizarse o incluso son posibles mejoras motoras clínicamente relevantes en adultos con 5q-SMA [6-8].
Congreso: DGM 2021
Literatura:
- Bowerman M, et al: Estrategias terapéuticas para la atrofia muscular espinal: SMN y más allá. Dis Model Mech 2017; 10: 943-954.
- Borasio G, et al: Diagnóstico de las atrofias musculares espinales. Neurología 2001; 20:113-118.
- www.sma-schweiz.ch/spinale-muskelatrophie/typen-der-proximalen-sma (último acceso 05.04.2021)
- www.muskelgesellschaft.ch/diagnosen/spinale-muskelatrophien-sma (último acceso 05.04.2021)
- Meyer, et al: Congreso DGN 2020, P333.
- Hagenacker T, et al: Lancet Neurol 2020; 19(4): 317-325.
- Walter MC, et al: J Neuromuscul Dis 2019; 6: 453-465.
- Maggi L, et al: JNNP. 2020, nov;91(11): 1166-1174.
InFo NEUROLOGY & PSYCHIATRY 2021; 19(3): 41 (publicado el 5.6.21, antes de impresión).
PRÁCTICA GP 2021; 16(8): 45