Las mejoras en el tratamiento de la enfermedad aumentan la calidad y la esperanza de vida de los pacientes con fibrosis quística. El cribado neonatal introducido en 2011 permite un diagnóstico precoz. Sin embargo, la enfermedad también debe considerarse como un posible diagnóstico diferencial en la edad adulta.
La fibrosis quística (FQ, a veces también fibrosis quística) es la enfermedad metabólica congénita, en última instancia mortal, más frecuente en Suiza. Como enfermedad sistémica, la FQ afecta principalmente a la función de las glándulas exocrinas del tracto respiratorio y del tracto digestivo. En particular, el curso pulmonar con destrucción continua e irreversible del órgano provoca el acortamiento de la esperanza de vida. Gracias a la mejora de las medidas médicas, los afectados llegan ahora a la edad adulta casi sin excepción, pero siguen representando un colectivo relativamente nuevo en la consulta del médico de cabecera. Como innovación prometedora, una terapia causal está disponible para una proporción muy pequeña de pacientes desde principios de 2014.
Patrón de la enfermedad
Ya en 1936, la fibrosis quística, que entonces aún tenía otro nombre, fue descrita por el pediatra zuriqués Guido Fanconi como una enfermedad mortal de los niños pequeños, razón por la cual su acceso permaneció cerrado a los colegas de la medicina de familia durante mucho tiempo.
La FQ es la enfermedad hereditaria congénita crónica y, en última instancia, limitante de la vida más común, con una prevalencia aproximada de 1:2500. Aproximadamente una de cada 25 personas en Europa Central es portadora de una mutación saludable. Hay unas 70.000 personas con la enfermedad en todo el mundo, y se calcula que unas 900 en Suiza. Aproximadamente la mitad de los pacientes tienen más de 18 años. El modo de herencia es autosómico recesivo, lo que significa que estadísticamente uno de cada cuatro hijos nacidos de una relación entre dos portadores sanos de la mutación está afectado por la FQ, dos hijos son también portadores sanos de la mutación como sus padres, y un hijo no es portador de la mutación ni está enfermo. En 1989, se identificó el gen subyacente en el cromosoma 7. La causa es una mutación en el gen CFTR (“regulador de la conductancia transmembrana de la fibrosis quística”), que codifica el canal de cloruro de la membrana celular [1,2].
Clínica
En la enfermedad sistémica se ven afectados diferentes órganos, pero normalmente la enfermedad pulmonar crónica conlleva un aumento de la morbilidad y una reducción de la esperanza de vida. Gracias a la mejora de las posibilidades médicas, la esperanza de vida ha aumentado continuamente en los últimos años. La esperanza media de vida en Europa supera actualmente los 40 años y seguirá aumentando, especialmente en la generación detectada en el cribado neonatal en Suiza desde 2011. Sin embargo, el curso de la enfermedad es muy inconsistente, y algunos pacientes siguen muriendo de insuficiencia pulmonar en la edad adulta temprana. Común a todos los enfermos son las infecciones bacterianas crónicas de las vías respiratorias, que conducen a la destrucción irreversible de los pulmones. Los pacientes sufren tos, expectoración y una creciente limitación de su capacidad de recuperación física. Además, la diarrea crónica con mala digestión se debe a la insuficiencia pancreática exocrina, que afecta a cerca del 85% de los pacientes. Como consecuencia de la malnutrición, pero también del aumento del trabajo respiratorio, se produce el retraso del crecimiento. Alrededor de un 10% ya presenta íleo meconial tras el nacimiento, pero las personas mayores también pueden sufrir síndromes de obstrucción intestinal recurrentes. A pesar de una terapia sintomática óptima, en el curso posterior de la enfermedad también puede producirse cirrosis hepática o insuficiencia pancreática endocrina con el desarrollo de diabetes mellitus. Los pacientes varones con FQ, en particular, suelen verse afectados por la infertilidad [1,2].
Fisiopatología
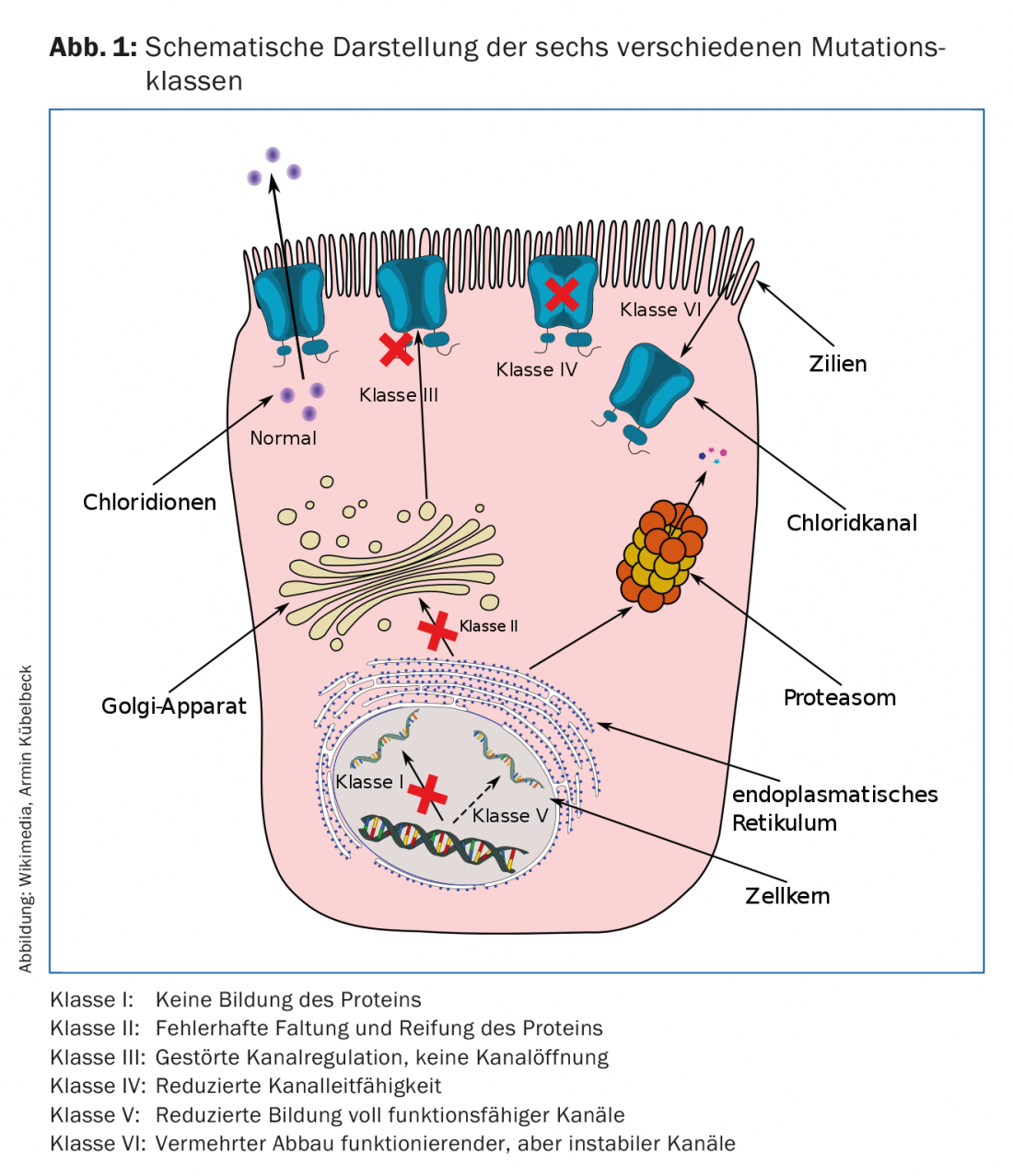
El gen CFTR del cromosoma 7 codifica el canal de cloruro de la membrana celular. Las mutaciones genéticas, de las que actualmente se conocen más de 2000 pero menos de 200 son claramente causantes de enfermedad, provocan un mal funcionamiento o la ausencia total del canal de cloruro en las células epiteliales. Las mutaciones del gen CFTR se dividen en seis clases (Fig. 1), que difieren en su patomecanismo. Algunas mutaciones, por ejemplo, provocan un fallo casi completo de la síntesis de la proteína CFTR. La mutación 3905insT, que se da en suizos o descendientes de suizos (por ejemplo, los amish en Norteamérica), se asigna a esta clase I y es la segunda mutación más frecuente en Suiza. En otras mutaciones, se impide la incorporación de la proteína a la membrana celular o el canal iónico de la proteína está bloqueado o sólo tiene una conductividad limitada. La mutación más frecuente en Europa, pero también en Suiza (86% de heterocigotos, 47% de homocigotos) se asigna a la clase II y se refiere a una deleción en la posición 508 (F508del). La proteína producida se pliega aquí incorrectamente y se degrada antes de asumir cualquier función. El fallo del canal de cloruro provoca la interrupción del transporte transepitelial en todos los órganos en los que las células epiteliales expresan CFTR. Tomando como ejemplo el epitelio de las vías respiratorias, se produce una cascada inflamatoria. El aumento de la viscosidad del moco provoca un deterioro del aclaramiento mucociliar, que a su vez se asocia a una colonización bacteriana precoz y, en el curso posterior, crónica. La respuesta inmunitaria del organismo provoca un reclutamiento excesivo de granulocitos neutrófilos, una liberación inadecuada de elastasa de neutrófilos y, en última instancia, la destrucción del tejido. Además de las dos mutaciones clásicas citadas como ejemplo (F508del, 3905insT), responsables de un curso grave de la enfermedad, ahora también se conocen mutaciones asociadas a un cuadro clínico más bien leve, mientras que en el caso de otras mutaciones no está claro si son siquiera causantes de la FQ. La base de datos www.cftr2.org intenta ofrecer una visión general continuamente actualizada de las mutaciones conocidas causantes de FQ y sus fenotipos probables [1,3,4].
Diagnóstico
Antes de 2011, alrededor del 10% de los niños con FQ eran diagnosticados de íleo meconial, pero la gran mayoría sólo eran identificados durante el curso de la enfermedad, cuando aparecían síntomas sospechosos de FQ. El patrón oro para el diagnóstico es la prueba del sudor, es decir, la determinación cuantitativa de la concentración de cloruro en el sudor tras la iontoforesis con pilocarpina. Además, la medición de la conductividad del sudor se utiliza a menudo como prueba de cribado, ya que esta prueba funciona de forma rápida y fiable con una cantidad significativamente menor de sudor. Macroduct® y Nanoduct® son sistemas de ensayo disponibles en el mercado. Para garantizar la calidad, la realización de pruebas de soldadura debe reservarse a los centros adecuados.
En 2011, la FQ se incluyó en el programa de cribado neonatal de Suiza. El tripsinógeno inmunorreactivo (IRT) se mide en la sangre del talón. Si se supera el límite, los recién nacidos son remitidos a un centro pediátrico de FQ. Allí se confirma o descarta el diagnóstico con una prueba del sudor y, si es necesario, exámenes complementarios (determinación de la elastasa pancreática, análisis genético).
En 2016, 143 niños habían sido diagnosticados de fibrosis quística a través del cribado neonatal. En un total estimado de 900 pacientes con fibrosis quística, alrededor del 15% de todos los pacientes podrían detectarse ya con la ayuda del cribado neonatal. Este valor seguirá aumentando de forma constante.
Sin embargo, es importante tener en cuenta que, debido a los diferentes fenotipos, puede haber casos diagnosticados en la infancia, así como aquellos con un curso leve o con una función CFTR residual medible. Por ello, es posible que sólo se manifieste en la edad adulta con síntomas atípicos como pancreatitis, sinusitis, pólipos nasales, bronquiectasias difusas y/o un deseo insatisfecho de tener hijos. Por lo tanto, el médico de cabecera también debe considerar la FQ como un posible diagnóstico diferencial en caso de que aparezcan los síntomas correspondientes [1,5–7].
Terapia y cuidados
Según las directrices internacionales, los pacientes deben ser conectados a centros especializados. Los pacientes son vistos a intervalos de 3 meses. Dependiendo de la evolución individual de la enfermedad, la terapia debe revisarse y ajustarse. Además de la historia clínica y el examen clínico, las mediciones de la función pulmonar (según la infraestructura, pletismografía corporal, espirometría, lavado de respiración múltiple N2) y las pruebas microbiológicas (frotis de esputo o faríngeo) forman parte de los exámenes rutinarios, que se complementan a intervalos mayores con imágenes de los pulmones y el abdomen, análisis de sangre, cargas de glucosa y mediciones de la densidad ósea. El objetivo es detectar los cambios en una fase temprana para poder contrarrestarlos y evitar un mayor deterioro.
El tratamiento básico es complejo e incluye una limpieza intensiva de las vías respiratorias mediante inhalación y fisioterapia respiratoria especial (solución salina hipertónica, rhDNAse si es necesario, “técnica de limpieza de las vías respiratorias” especial), sustitución de enzimas pancreáticas (lipasa) y terapia nutricional (sustitución de vitaminas liposolubles, dieta hipercalórica), así como un tratamiento antimicrobiano agresivo (inhalación y/o aplicación sistémica).
Dependiendo de la edad del paciente y de las circunstancias que le acompañen, también es necesario un asesoramiento continuo y específico para poder aceptar de forma óptima los retos de la enfermedad y sus efectos en la forma de vida. Además de los contenidos obvios relacionados con la enfermedad y la terapia, los pacientes (y dependiendo de su edad, también sus tutores legales) también necesitan apoyo para hacer frente a cuestiones sociales, psicológicas, financieras y relacionadas con los seguros (discapacidad y/o seguro médico), para organizar el cuidado de los niños, la asistencia a guarderías y escuelas, la formación y el trabajo, los viajes, el deseo de los padres de volver a tener hijos o la propia planificación familiar del paciente adulto, etc.
También hay que tener en cuenta que, afortunadamente, los afectados son cada vez más mayores. De repente, los pacientes, pero también los equipos que los tratan, están expuestos a problemas internos que surgen independientemente de la FQ pero que influyen en su curso. Para responder a estas demandas, es necesario un enfoque terapéutico multidisciplinar, que sólo puede proporcionar un centro de FQ [8,9].
Outlook
En 2014, Swissmedic aprobó el fármaco ivacaftor (Kalydeco®) para pacientes con la mutación G551D de clase III. Por primera vez, una terapia causal con un aumento significativo de la función del canal de cloruro, medible en una mejora notable de la prueba del sudor y de la función pulmonar, podría utilizarse en los pocos pacientes con esta mutación (aproximadamente el 4-5% de todos los pacientes del mundo). Actualmente se están probando otros moduladores del CFTR. Queda la esperanza de que una terapia causal específica para la mutación esté disponible para un grupo más amplio de pacientes en el futuro [10].
Mensajes para llevarse a casa
- Gracias a las mejoras en la terapia sintomática y el tratamiento de la enfermedad, la calidad y la esperanza de vida aumentan continuamente.
- Hoy en día, la mitad de los pacientes suizos de fibrosis quística son adultos.
- El cribado neonatal de la fibrosis quística introducido en 2011 permite un diagnóstico precoz. Esto significa que la terapia básica puede iniciarse antes de que se produzcan los primeros cambios relacionados con la enfermedad.
- Son posibles los cursos oligosintomáticos y atípicos. Por lo tanto, la FQ también debe considerarse como un posible diagnóstico diferencial en la edad adulta.
- Las aclaraciones y los cuidados posteriores deben realizarse siempre en colaboración con un centro de FQ.
Literatura:
- Elborn JS: Fibrosis quística. Lancet 2016 Nov 19; 388(10059): 2519-2531.
- Zolin A, et al: Informe anual de la ECFSPR 2014. 2016.
- Hergersberg M, et al.: Una nueva mutación, 3905insT, representa el 4,8% de 1173 cromosomas de FQ en Suiza y causa un fenotipo grave. Hum Genet 1997 Ago; 100(2): 220-223.
- CFTR2. www.cftr2.org
- Torresani T, et al: Cribado de la fibrosis quística en recién nacidos en Suiza: consecuencias tras el análisis de un estudio piloto de 4 meses. J Cyst Fibros 2013; 12(6): 667-674.
- Cribado neonatal Suiza. www.neoscreening.ch
- Barben J, et al: Cribado neonatal de la fibrosis quística: una historia de éxito. Schweiz Med Forum 2013; 13(49): 1010-1012.
- Smyth AR, et al: Normas de atención de la Sociedad Europea de Fibrosis Quística: directrices de buenas prácticas. J Cyst Fibros 2014 mayo; 13(Suppl 1): S23-42.
- Elborn JS, et al: Informe del grupo de trabajo de la Sociedad Respiratoria Europea/Sociedad Europea de Fibrosis Quística sobre la atención a adultos con fibrosis quística. Eur Respir J 2016 Feb; 47(2): 420-428.
- Ramsey BW, et al: Un potenciador CFTR en pacientes con fibrosis quística y la mutación G551D. N Engl J Med 2011 Nov 3; 365(18): 1663-1672.
PRÁCTICA GP 2017; 12(11): 31-33