Una linfadenopatía persistente es una indicación clara para realizar diagnósticos histopatológicos adicionales. Un historial médico detallado, así como el conocimiento de enfermedades previas, manipulaciones y terapias farmacológicas son esenciales para un diagnóstico definitivo del linfoma. Hay casos que se encuentran en zonas grises en términos de clasificación. La presencia de reordenamientos MYC es un biomarcador pronóstico indiscutible en el DLBCL, al igual que la coexpresión fenotípica de las proteínas myc y bcl2. La respuesta a la terapia con rituximab está relacionada con la expresión de CD20. Los estudios moleculares están aportando nuevos parámetros genéticos predictivos de la respuesta específica a las terapias dirigidas en el linfoma.
Con una cuota relativa del 5%, los linfomas representan el quinto grupo más frecuente de enfermedades malignas en ambos sexos. Especialmente los linfomas de células B maduras (antes “linfomas no Hodgkin”) muestran el mayor crecimiento de incidencia de malignomas en el mundo industrializado después del melanoma.
Se desconocen las razones, pero podrían estar relacionadas con el aumento de la esperanza de vida, la creciente incidencia de enfermedades autoinmunes y el uso generalizado asociado de (nuevos) inmunosupresores, o la creciente exposición a ciertos pesticidas y herbicidas. La incidencia del linfoma se sitúa actualmente en torno a los 25 casos/100.000 habitantes/año.
El progreso de la terapia oncológica es evidente en el caso de los linfomas. Las tasas de supervivencia a 5 años específicas de la enfermedad para los linfomas ganglionares más comunes, como el linfoma difuso de células B grandes (LDCBG), el linfoma folicular (LF), el linfoma de Hodgkin clásico (LH) y el linfoma linfoblástico de células B (LBL-B, equivalente ganglionar de la leucemia linfoblástica aguda de células B), se sitúan en torno al 60% (LDCBG) y alrededor del 80% (el resto de entidades). Esto explica la elevada prevalencia del linfoma.
Definición
Los linfomas se definen como enfermedades neoplásicas malignas de linfocitos B, T o NK inmaduros o maduros en órganos del sistema linfático (nodales) o fuera de dichos órganos (extranodales). Pueden ser leucémicos o sin exudación (linfomas en sentido estricto).
Clínica
Los linfomas ganglionares se presentan con linfadenopatía localizada o generalizada, persistente (más de tres semanas), a menudo progresiva, con o sin síntomas generales (B) (fiebre, sudores nocturnos, pérdida de peso), afectación de órganos, cambios cutáneos (prurito, eritema) o signos de insuficiencia de la médula ósea (anemia, petequias, tendencia a la infección).
Por lo tanto, una linfadenopatía persistente, especialmente acompañada de los síntomas mencionados, es una clara indicación para un diagnóstico más profundo.
Diagnóstico
Los diagnósticos basados en tejidos son indispensables en los linfomas, ya que los cambios histopatológicos de los tejidos son la piedra angular de la dignidad y la determinación de la entidad. Basándose en estos cambios, es posible distinguir entre benigno y maligno y el grado de maduración de los linfocitos afectados mediante microscopía óptica convencional. Mediante otros métodos microscópicos in situ, como la inmunohistoquímica (expresión de proteínas) o la hibridación fluorescente in situ (aberraciones cromosómicas recurrentes), puede determinarse la filiación de linaje (linaje B, T o NK), el estadio exacto de desarrollo (por ejemplo, célula B de centro germinal) y las expresiones de marcadores patológicos (por ejemplo, expresión de marcadores de células T en células B, como en la leucemia linfocítica crónica de células B). [B-CLL]) o una translocación cromosómica existente (por ejemplo, t[14;18] en el FL) puede determinarse y hacer un diagnóstico exacto. Se trata de un requisito previo básico para una terapia oncológica específica. En los casos difíciles desde el punto de vista del diagnóstico, puede obtenerse ADN del material (fijado en formol e incluido en parafina) y analizarse más a fondo en busca de clonalidad de células B y T, translocaciones y mutaciones puntuales.
Clasificación
Los linfomas se clasifican según la clasificación actual de la OMS. El principio rector es el diagnóstico integrador, que considera de igual importancia a la hora de clasificar las entidades la inclusión de la morfología histopatológica del linfoma, los fenotipos (patrón de expresión proteica), los genotipos (aberraciones cromosómicas recurrentes) y la clínica.
Se ha abandonado la separación primaria de los linfomas en linfomas Hodgkin y no Hodgkin. Debido a su morfología característica, su presentación clínica y su excelente respuesta a terapias específicas, el LH sigue gestionándose como una entidad diferenciada.
Nuevas categorías: Debido a la complejidad biológica de ciertas enfermedades, la OMS ha introducido dos categorías denominadas “zona gris”:
- Linfomas de células B inclasificables con características intermedias entre un LDCBG y un LH
- Linfomas de células B inclasificables con características intermedias entre un LDCBG y un linfoma de Burkitt (BL).
Aparte del solapamiento clínico y morfológico entre las entidades individuales de ambas categorías, los estudios moleculares muestran un solapamiento significativo de los genes expresados entre los linfomas mediastínicos primarios de células B grandes y el LH, por un lado, y entre el LDCB individual y el BL, por otro.
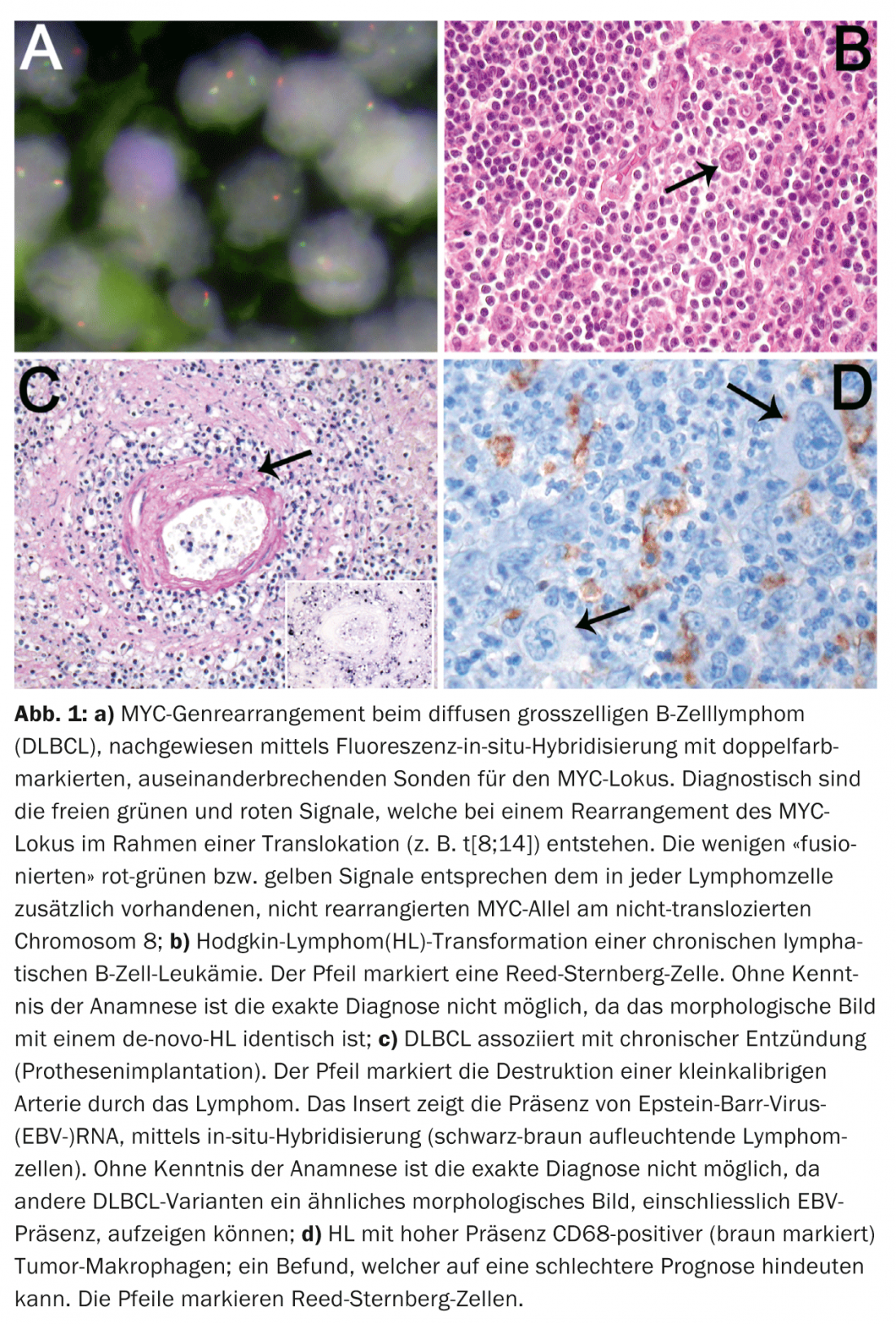
La evolución clínica desfavorable de los casos de LDCBG altamente proliferativos con disposición del gen MYC (Fig. 1a ) justifica aún más la introducción de esta categoría de zona gris para el seguimiento de estos casos, que presentan una respuesta inadecuada a las terapias estándar de LDCBG, en ensayos terapéuticos prospectivos.
Categorías diagnósticas basadas en parámetros clínicos: En la clasificación de la OMS se tuvo muy en cuenta el contexto relacionado con el paciente, como la edad, el tratamiento farmacológico previo o en curso, especialmente el inmunosupresor, y la localización. Se definen cuatro entidades en función de la edad de los pacientes:
- FL pediátrica
- Linfoma pediátrico de células B de la zona marginal ganglionar (LBZM)
- Enfermedad linfoproliferativa de células T de la infancia asociada al virus de Epstein-Barr (VEB)
- LDCBG asociado al VEB en ancianos.
La justificación de la introducción de estas entidades es la asociación de la aparición con la inmadurez o senescencia del sistema inmunitario. Mientras que los tres primeros enumerados son claramente raros, la proporción relativa de los últimos es del 3% de todos los DLBCL. Aunque los estudios del Lejano Oriente indican una clara agresividad de este linfoma, nuestros datos de Europa Central muestran una agresividad clínica particular sólo en casos individuales.
Como reflejo de la frecuencia y heterogeneidad del DLBCL, se han definido variantes clinicopatológicas adicionales en la nueva clasificación. Para algunas de estas variantes, como el DLBCL asociado a una inflamación crónica (piotórax, osteomielitis crónica, implantes de cuerpos extraños infectados, es decir, prótesis vasculares/articulares, úlceras cutáneas crónicas, articulaciones artrósicas en la artritis reumatoide), el conocimiento de la clínica es indispensable en términos de clasificación. (Fig. 1b). Dado que el 70% de estos linfomas están asociados al VEB y se dan sobre todo en pacientes de edad avanzada, la diferenciación del DLBCL asociado al VEB en ancianos sólo es posible sobre la base de pruebas anamnésicas.
El conocimiento de la clínica también es indispensable para el diagnóstico de las enfermedades linfoproliferativas iatrogénicas, asociadas a inmunodeficiencias (terapia con inmunosupresores), así como para el diagnóstico de las linfoproliferaciones postrasplante (trasplante de órganos o de médula ósea alogénica). Lo mismo se aplica al diagnóstico de los “linfomas de células B pequeñas” secundarios, transformados (LLC-B, LZM, LF), que pueden transformarse en un LDCB, en un linfoma de células B inclasificable con características intermedias entre un LDCB y un LB, así como en un LH. La indicación anamnésica de un linfoma indolente previo del grupo de los “linfomas de células B pequeñas” es decisiva para la correcta clasificación de dichas lesiones. Esto es significativo porque el LDCB o el LH (Fig. 1c), que se transforman a partir de dichos linfomas de células B, tienen un curso significativamente más agresivo que sus análogos de novo.
Pronóstico y predicción
Numerosos estudios realizados en la última década han tratado de establecer factores pronósticos significativos relacionados con el tumor en los linfomas ganglionares más comunes, como el LDCBG, el LF y el LH. Sin embargo, los factores pronósticos clínicos conocidos, como el Índice Pronóstico Internacional (IPI) en el LDCBG, el Índice Pronóstico Internacional FL (FLIPI) y la Puntuación Pronóstica Internacional (IPS) en el LH, no se superaron al incluir factores pronósticos relacionados con el tumor. A excepción de unas pocas variantes de DLBCL que se asocian a un pronóstico bastante peor, como el DLBCL rico en células T e histocitos, la granulomatosis linfomatoide y el DLBCL intravascular, sólo la detección de reordenamientos MYC es un parámetro pronóstico indiscutible relacionado con el tumor en el DLBCL. Tres nuevos grandes estudios independientes han demostrado el papel pronóstico desfavorable de la coexpresión fenotípica de las proteínas myc y bcl2 (el denominado “DLBCL de doble impacto fenotípico”). Los estudios en HL y FL mostraron un efecto pronóstico de la composición de células T de fondo. Estudios recientes de expresión génica también han demostrado que las firmas de los macrófagos asociados al tumor pueden influir significativamente en la supervivencia en el LH, como reflejan morfológicamente los altos niveles de macrófagos tisulares en pacientes con pronóstico desfavorable (Fig. 1d).
En resumen, los marcadores pronósticos en el linfoma aún no están listos para la práctica diaria. Sin embargo, la búsqueda de parámetros pronósticos asociados al microentorno tumoral parece prometedora, sobre todo porque este entorno también podría manipularse terapéuticamente sin temor a que el tumor desarrolle resistencia.
Otro campo de investigación se refiere al establecimiento de marcadores predictivos, es decir, biomarcadores que indiquen la respuesta o la falta de respuesta a una terapia. Aunque los ensayos clínicos no han investigado específicamente la expresión de CD20 en el establecimiento del anticuerpo terapéutico anti-CD20 rituximab, la experiencia demuestra que sólo los linfomas de células B que expresan CD20 responden a esta terapia. La determinación de la expresión de CD20 en los linfomas es, por tanto, un ejemplo de marcador predictivo determinable histopatológicamente.
Datos recientes sugieren que la sensibilidad específica a los inhibidores de la cinasa de Bruton (por ejemplo, el ibrutinib), los agentes promotores de la apoptosis (por ejemplo, el obatoclax) y los inhibidores de la fosfoinositida 3-cinasa (incluidos los inhibidores de mTOR como el everolimus) puede predecirse a partir de cambios genéticos específicos en las células linfoides.
Prof. Dr. Alexandar Tzankov
Prof. Dr. med. Stephan Dirnhofer
Literatura:
- Roman E, Smith AG: Histopatología 2011; 58: 4-14.
- Swerdlow SH, et al: Clasificación de la OMS de los tumores de los tejidos hematopoyéticos y linfoides. Lyon: CIIC; 2008.
- Hoeller S, et al: Hum Pathol 2010; 41: 352-357.
- Tzankov A, et al.: Mod Pathol 2013; doi: 10.1038/modpathol.2013
- Hu S, et al: Blood 2013; 121: 4021-4031.
- Steidl C, et al: N Engl J Med 2010; 362: 875-885.
- Tzankov A, et al: Haematologica 2008; 93: 193-200.
- Rahal R, et al: Nat Med 2014; 20: 87-92.
- Wenzel SS, et al: Leucemia 2013; 27: 1381-1390.
- Pfeifer M, et al: Proc Natl Acad Sci U S A. 2013; 110: 12420-12425.
InFo Oncología y Hematología 2014; 2(2): 5-7