La destrucción crónica de los glóbulos rojos es característica de la hemoglobinuria paroxística nocturna. Las mutaciones en el gen PIG-A provocan un déficit de GPI-AP, que da lugar a una activación incontrolada del complemento. La inhibición del complemento C5, que sólo debe administrarse cada ocho semanas por primera vez, puede ayudar.
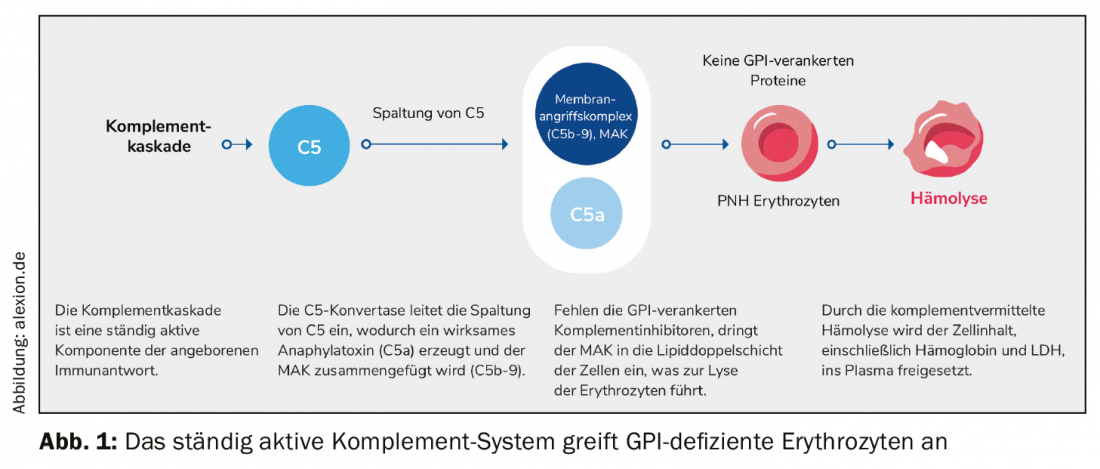
La característica principal de la hemoglobinuria paroxística nocturna (HPN), rara, crónica y progresiva, es la hemólisis mediada por complemento. Debido a una mutación adquirida, faltan proteínas de anclaje del glicofosfatidilinostiol (GPI) en las células madre hematopoyéticas de la médula ósea. Como resultado, no se pueden formar proteínas ancladas a GPI, especialmente en la superficie de los eritrocitos rojos. La protección contra el sistema del complemento, una parte del propio sistema inmunitario del organismo, desaparece. Los glóbulos rojos son confundidos con invasores, atacados y destruidos. El complejo de ataque a la membrana (MAK) penetra en la bicapa lipídica de la célula y desencadena allí la lisis de los eritrocitos. Todo el contenido celular, incluida la hemoglobina y la LDH, se libera al plasma (Fig. 1).
Enfermedad compleja con síntomas variables
La enfermedad se caracteriza por anemia hemolítica, hemoglobinuria e insuficiencia o fallo de la médula ósea, entre otros síntomas. Clásicamente se manifiesta sobre todo por fatiga, disminución de la calidad de vida y disnea de esfuerzo. Sin embargo, también pueden aparecer los síntomas inespecíficos que suelen acompañar a las crisis hemolíticas. Sin embargo, la principal causa del aumento de la morbilidad y la mortalidad de los pacientes es la trombofilia. Hasta un 50% de todos los afectados desarrollan trombosis sin medidas terapéuticas específicas. El 35% de los pacientes fallece en los 5 años siguientes al diagnóstico.
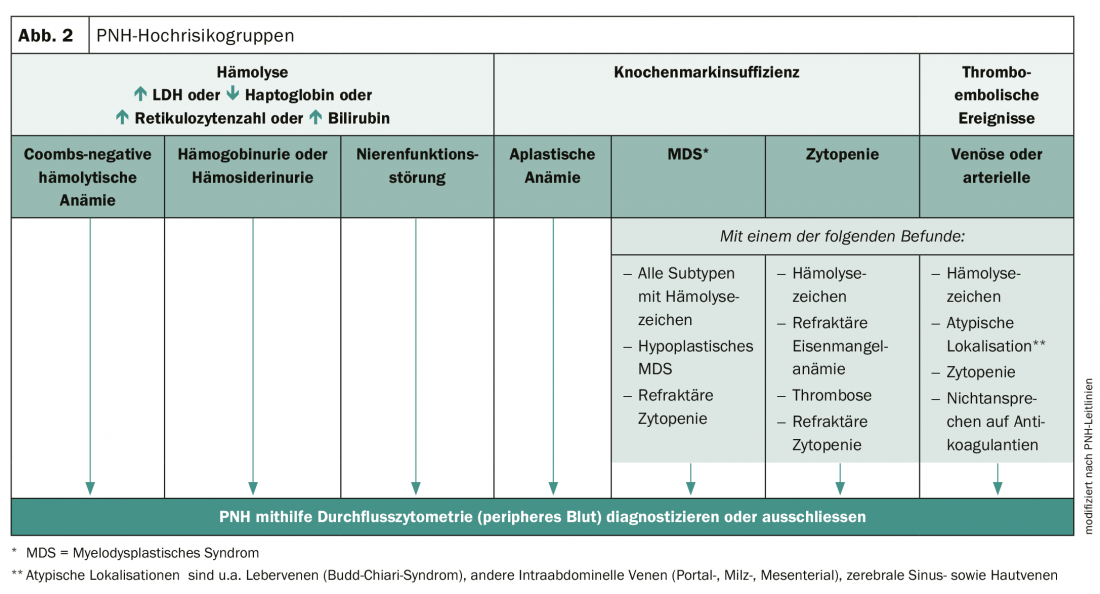
El diagnóstico precoz es esencial para mejorar el pronóstico de los afectados. La HPN puede diagnosticarse mediante citometría de flujo de alta sensibilidad y un examen clínico exhaustivo. Sin embargo, muchos médicos no son conscientes de la complejidad de los síntomas y la variabilidad de la enfermedad. Como consecuencia, la HPN no suele reconocerse y el diagnóstico sólo se realiza tras un retraso de uno a más de 5 años. Sin embargo, existen grupos de alto riesgo en los que debe considerarse la presencia de HPN (Fig. 2).
La gestión de la terapia mejora
Para detener la hemólisis, se interfiere en las funciones terminales de la cascada del complemento mediante inhibidores del complemento. Las funciones proximales, en cambio, permanecen. Por ejemplo, la tasa de acontecimientos tromboembólicos (ET) se redujo significativamente de 11,54 a 0,72 en los pacientes tratados con anticoagulantes. El inhibidor también resultó convincente en términos de beneficio clínico a largo plazo. El único inconveniente: el corto intervalo de infusión. Ahora se podría seguir desarrollando el inhibidor y prolongar su vida media (Ravulizumab). Al reducir el número de infusiones a 6-7 al año, la calidad de vida de los afectados mejora significativamente y el riesgo de hemólisis intercurrente también se ha reducido del 10,7% al 4%.
Fuente: Reunión Anual 2019 de las Sociedades de Habla Alemana de Hematología y Oncología Médica (DGHO)
InFo ONCOLOGY & HEMATOLOGY 2019; 7(6): 32-33 (publicado el 6.12.19, antes de impresión).