El tratamiento quirúrgico de los pacientes con enfermedades hereditarias del tejido conjuntivo tiene como objetivo prevenir la disección aórtica. Porque esto es responsable de la elevada tasa de mortalidad de la población de pacientes. ¿Cuándo se da la indicación? ¿Y qué opciones de tratamiento farmacológico existen?
Las enfermedades hereditarias del tejido conjuntivo con manifestaciones vasculares apenas están presentes en la conciencia del médico general. Durante mucho tiempo, el síndrome de Marfan fue el único diagnóstico diferencial en este sentido en pacientes jóvenes con un evento aórtico torácico. En la última década se han identificado cientos de genes asociados a formas sindrómicas y no sindrómicas de la enfermedad aórtica [1].
Sin embargo, casi la mitad de los pacientes sólo se diagnostican en el contexto de una complicación vascular. Mientras que los aneurismas suelen detectarse durante un examen rutinario, la disección aórtica es una de las urgencias quirúrgicas asociadas a una elevada morbilidad y mortalidad.
El síndrome de Marfan es un trastorno autosómico dominante con una incidencia aproximada de 1:5000 nacidos vivos. Las manifestaciones típicas oculares, esqueléticas y cardíacas están causadas por mutaciones en el gen de la fibrilina-1, que conducen a la sobreactivación de la vía de señalización del TGFβ [2].
Bart Loeys y Hal Dietz identificaron hace diez años una subpoblación de pacientes que llamaban la atención por su úvula partida, su amplia distancia interocular y sus vasos tortuosos. Mientras tanto, se han identificado varios genes que conducen a un fenotipo del grupo del síndrome de Loeys-Dietz. Identificar a estos pacientes es importante porque las disecciones suelen producirse en diámetros aórticos que antes no se consideraban indicaciones de sustitución aórtica profiláctica [3].
El raro síndrome vascular de Ehlers-Danlos está causado por mutaciones en el gen que codifica el colágeno III. Estos pacientes destacan por su elevada tasa de disección y rotura sin formación previa de aneurisma. Esto hace que el cuidado de estos pacientes sea muy difícil. La supervivencia media es de 48 años si no se trata, y los primeros episodios suelen producirse en la tercera o cuarta década de la vida [4].
Opciones de tratamiento quirúrgico
El objetivo del tratamiento quirúrgico de los pacientes con enfermedades hereditarias del tejido conectivo es prevenir la disección aórtica debida a la dilatación de la aorta, ya que es la responsable de la elevada mortalidad en esta población de pacientes. Las directrices de la Sociedad Europea de Cardiología (ESC) de 2014 hacen una clara distinción en la indicación de la sustitución aórtica electiva entre los pacientes de Marfan o Loeys-Dietz y los pacientes sin enfermedad genética subyacente [5]. La dificultad reside aquí en la delimitación. En pacientes jóvenes, debe suponerse la presencia de un componente genético. En pacientes sin enfermedad del tejido conjuntivo, se recomienda la sustitución aórtica profiláctica cuando el diámetro de la raíz aórtica o de la aorta ascendente sea de 55 mm. Los pacientes de Marfan, por su parte, deben ser operados a partir de 50 mm, y en el caso de que existan factores de riesgo (como disecciones en la familia) incluso a partir de 45 mm. En los pacientes Loeys-Dietz, se aconseja la sustitución electiva a los 42-45 mm. Por regla general, se sustituye principalmente la raíz aórtica. Si es posible, se intenta conservar la propia válvula del paciente. Si esto no es factible debido a daños en la válvula, se implanta un conducto valvulado.
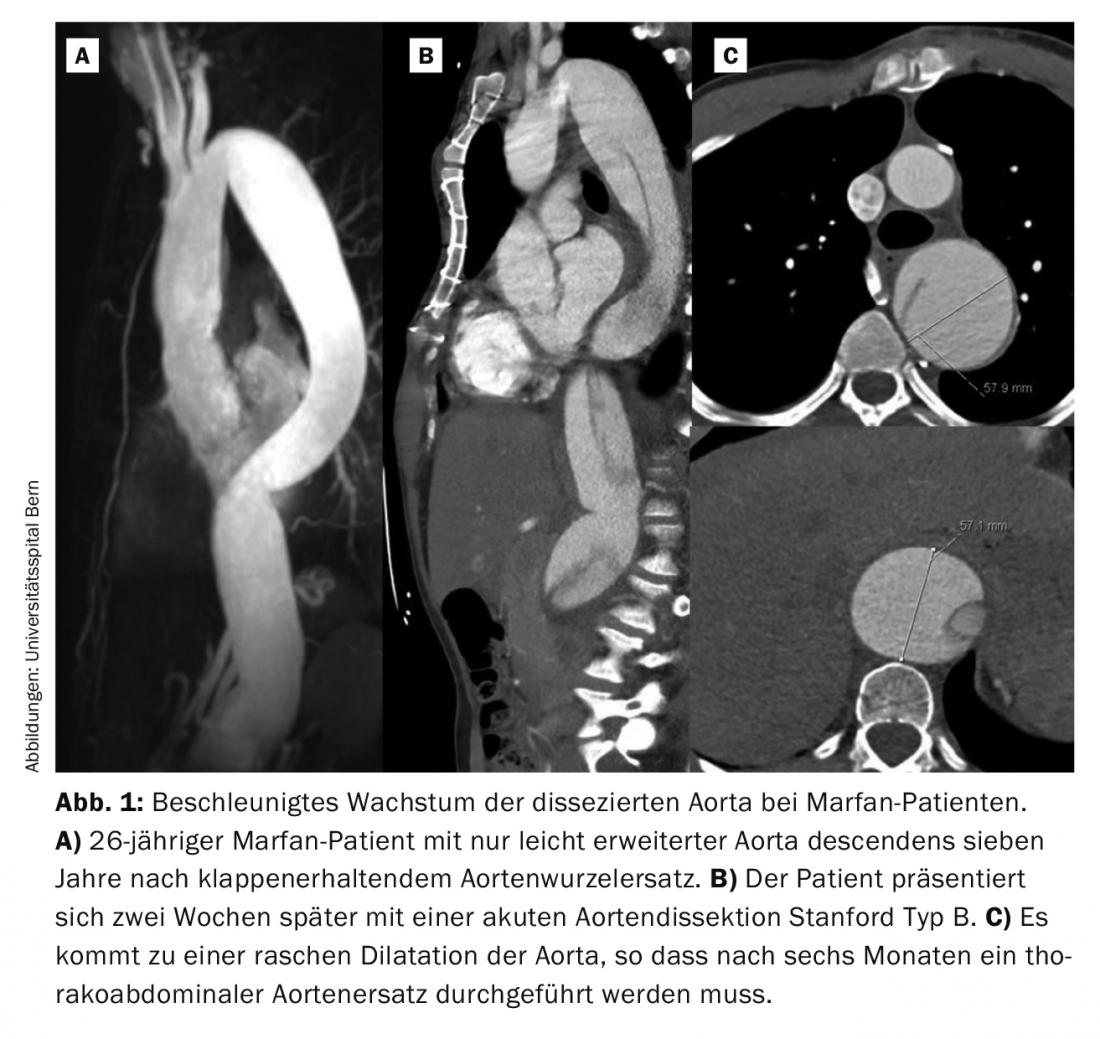
Sin embargo, a menudo la manifestación inicial es una disección aórtica aguda tipo Stanford A o B. En nuestra experiencia de los últimos veinte años, un tercio de los pacientes presentaban una disección aguda. La disección aguda de tipo A es una urgencia quirúrgica y debe tratarse inmediatamente. Incluso con una intervención quirúrgica exitosa, la mitad de estos pacientes requieren una reintervención, sobre todo en la aorta distal, es decir, la no reemplazada. En los pacientes con sustitución electiva de la raíz aórtica, sólo un 10% de los pacientes necesitan repetir la operación. En la mayoría de los casos, se trata de pacientes que han sufrido entretanto una disección aórtica de tipo B. Aunque la disección aórtica de tipo B suele ser inicialmente “no complicada”, es decir, sin malperfusión, en la gran mayoría de los pacientes de Marfan la aorta debe sustituirse posteriormente de forma toracoabdominal. Una característica típica de los pacientes de Marfan es el rápido crecimiento de la aorta disecada en las primeras semanas y unos pocos meses (Fig. 1) . La experiencia demuestra que el 50% de los pacientes requieren cirugía en el primer año tras el suceso [6].
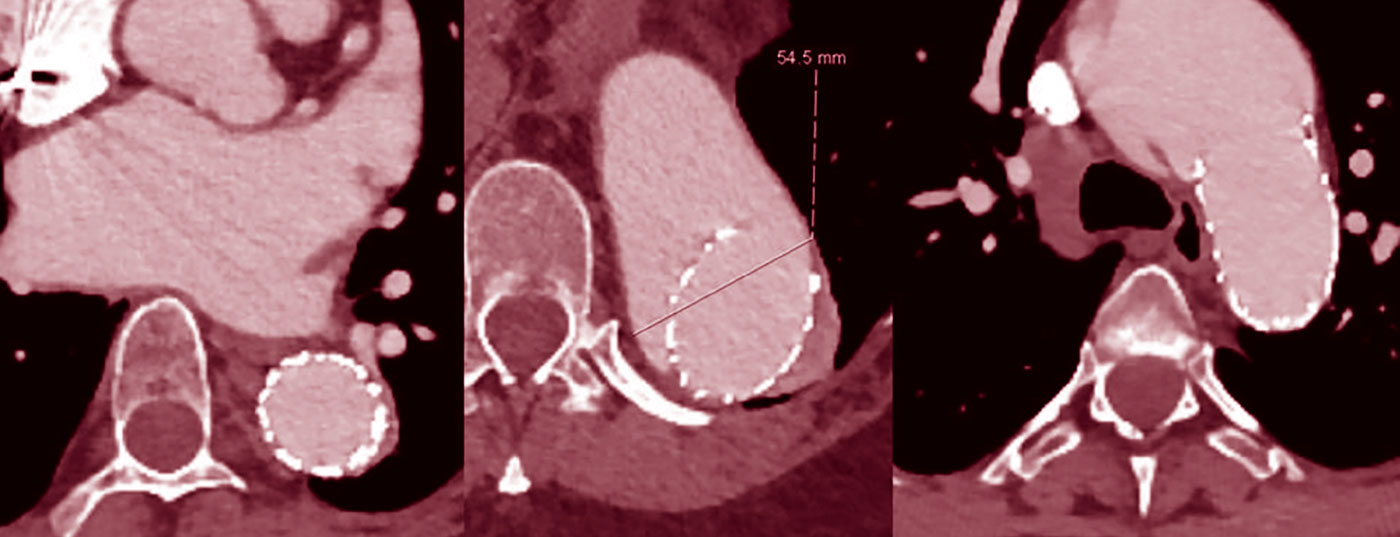
Por lo general, no se recomienda el tratamiento de los segmentos aórticos toracoabdominales dilatados con injertos de endoprótesis porque los segmentos de los vasos de la zona de aterrizaje suelen dilatarse secundariamente (Fig. 2), aunque inicialmente haya una buena remodelación [7].

Opciones de medicamentos
La dilatación y el consiguiente riesgo de disección de la aorta es la principal causa de morbilidad y mortalidad en pacientes con enfermedades hereditarias del tejido conectivo. El objetivo del tratamiento farmacológico es reducir la progresión de la dilatación y la aparición de disecciones. La reducción de la presión arterial a valores sistólicos máximos de 120 mmHg y el ritmo de aumento de la presión en la región aórtica con cada latido son de importancia relevante. Tradicionalmente, el betabloqueante es el fármaco de elección [8]. Los estudios han demostrado una menor tasa de dilatación en pacientes en tratamiento con betabloqueantes, pero sin significación clínica en términos de supervivencia [9].
En los últimos años, se ha estudiado con más detalle el bloqueante de los receptores ATII losartán. Dado que interfiere directamente en la vía de señalización del TGFβ, se han depositado grandes esperanzas en su efecto con respecto a la prevención de la dilatación y la disección aórticas [10]. Sin embargo, un gran ensayo aleatorizado en niños con síndrome de Marfan no encontró diferencias entre los betabloqueantes y el losartán [11]. El estudio fue controvertido porque en última instancia comparaba una dosis muy alta de atenolol con una dosis relativamente baja de losartán. Debido a su menor perfil de efectos secundarios, el antagonista ATII es, por tanto, nuestra primera elección para el tratamiento de pacientes sin tratamiento y con una función de bomba normal. Los antagonistas ATII son bien tolerados, especialmente por niños y adolescentes. Estudios más pequeños han demostrado que la presión arterial no se reduce significativamente en esta población de pacientes [12].
Controles de progreso
Para detectar a tiempo la aparición o progresión de un aneurisma aórtico y evitar la disección aórtica mediante una sustitución aórtica profiláctica precoz, es esencial llevar a cabo un estrecho seguimiento [13]. Es importante evaluar sistemáticamente todos los segmentos aórticos. Además, los controles tras la cirugía aórtica son importantes para detectar precozmente posibles complicaciones como la formación de aneurismas en otras secciones de la aorta y disecciones asintomáticas.
En nuestras horas de consulta, los pacientes son revisados postoperatoriamente a los tres y doce meses mediante angio-TC y posteriormente, en función de los hallazgos, una o tres veces al año. Para reducir la dosis de radiación acumulada, ésta debe realizarse principalmente mediante resonancia magnética. El examen por IRM también ofrece la ventaja de la imagen funcional con respecto a la función de la bomba, la vitiación de la válvula y el diagnóstico de isquemia. Las operaciones previas con implantes rara vez suponen un problema a la hora de evaluar las imágenes. Actualmente, la angiografía por TC sigue siendo superior a la RM en la evaluación de las disecciones. Se realizan controles ecocardiográficos en caso de vitia o st.n. Reemplazo valvular realizado una vez al año o alternativamente con examen de resonancia magnética.
Una situación especial en el cuidado a largo plazo de pacientes con enfermedades hereditarias del tejido conectivo es el deseo de tener hijos. Existen pocas pruebas al respecto y las recomendaciones de las sociedades profesionales son en parte contradictorias [5,14,15]. En general, un diámetro aórtico de <40 mm se considera un riesgo aceptable de quedarse embarazada bajo estrecho control ecocardiográfico y bloqueo beta. Para los diámetros >45 mm, se aconseja claramente la cirugía profiláctica. Para diámetros entre 40 y 45 mm, debe sopesarse muy cuidadosamente la situación individual del paciente. El factor de riesgo en este caso es sin duda un historial familiar positivo en cuanto a disecciones, especialmente durante el embarazo.
Las directrices actuales recomiendan el cribado de todos los familiares de primer grado de un paciente con aneurisma de aorta torácica. Este paso puede contribuir significativamente a reducir la tasa de pacientes con disecciones.
Mensajes para llevarse a casa
- En pacientes con enfermedades hereditarias del tejido conectivo, la indicación de una sustitución aórtica profiláctica se da ya en diámetros de 45-50 mm.
- La evaluación regular y sistemática de todos los segmentos aórticos previene las disecciones y las roturas.
- Todos los familiares de primer grado de pacientes con un aneurisma de aorta torácica deben someterse a un cribado para detectar la presencia de uno.
Literatura:
- Brownstein AJ, et al: Genes asociados al aneurisma y la disección de la aorta torácica: actualización de 2018 e implicaciones clínicas. Aorta (Stamford) 2018; 6: 13-20.
- Habashi JP, et al: El losartán, un antagonista AT1, previene el aneurisma aórtico en un modelo de ratón del síndrome de Marfan. Science 2006; 312: 117-121.
- Loeys BL, et al: Síndromes de aneurisma causados por mutaciones en el receptor TGF-beta. N Engl J Med 2006; 355: 788-798.
- Pepin M, et al: Características clínicas y genéticas del síndrome de Ehlers-Danlos tipo IV, el tipo vascular. N Engl J Med 2000; 342: 673-680.
- Erbel R, et al.: 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. El Grupo de Trabajo para el Diagnóstico y Tratamiento de las Enfermedades Aórticas de la Sociedad Europea de Cardiología (ESC). Eur Heart J 2014; 35: 2873-2926.
- Schoenhoff FS, et al: La disección aórtica aguda determina el destino de los segmentos aórticos no tratados inicialmente en el síndrome de Marfan. Circulation 2013; 127: 1569-1575.
- Grabenwöger M, et al: Reparación aórtica endovascular torácica (TEVAR) para el tratamiento de las enfermedades aórticas: declaración de posición de la Asociación Europea de Cirugía Cardio-Torácica (EACTS) y la Sociedad Europea de Cardiología (ESC), en colaboración con la Asociación Europea de Intervenciones Cardiovasculares Percutáneas (EAPCI). Eur Heart J 2012; 33: 1558-1563.
- Shores J, et al: Progresión de la dilatación aórtica y beneficio del bloqueo betaadrenérgico a largo plazo en el síndrome de Marfan. N Engl J Med 1994; 330: 1335-1341.
- Gersony DR, et al: El efecto del tratamiento con betabloqueantes en los resultados clínicos de los pacientes con síndrome de Marfan: un metaanálisis. Int J Cardiol 2007; 114: 303-308.
- Habashi JP, et al.: La señalización del receptor tipo 2 de la angiotensina II atenúa el aneurisma aórtico en ratones mediante el antagonismo de ERK. Science 2011; 332: 361-365.
- Lacro RV, et al: Atenolol frente a losartán en niños y adultos jóvenes con síndrome de Marfan. N Engl J Med 2014; 371: 2061-2071.
- Brooke BS, et al: Bloqueo de la angiotensina II y dilatación de la raíz aórtica en el síndrome de Marfan. N Engl J Med 2008; 358: 2787-2795.
- Jondeau G, et al: Aortic event rate in the marfan population a cohort study. Circulation 2012; 125: 226-232.
- Baumgartner H, et al: Directrices de la ESC para el tratamiento de las cardiopatías congénitas del adulto (nueva versión 2010). Eur Heart J 2010; 31: 2915-2957.
- Hiratzka LF, et al.Directrices 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM para el diagnóstico y tratamiento de pacientes con enfermedad aórtica torácica: un informe del Grupo de Trabajo sobre Directrices Prácticas de la Fundación del Colegio Americano de Cardiología/Asociación Americana del Corazón, la Asociación Americana de Cirugía Torácica, el Colegio Americano de Radiología, la Asociación Americana de Accidentes Cerebrovasculares, la Sociedad de Anestesiólogos Cardiovasculares, la Sociedad de Angiografía e Intervenciones Cardiovasculares, la Sociedad de Radiología Intervencionista, la Sociedad de Cirujanos Torácicos y la Sociedad de Medicina Vascular. Circulation 2010; 121: e266-369.
CARDIOVASC 2018; 17(5): 27-29









