Las dermatosis bullosas autoinmunes son un grupo clínica e inmunopatológicamente heterogéneo de enfermedades autoinmunes raras. En los pacientes ancianos con prurito y ampollas abultadas, el penfigoide bulloso encabeza la lista de diagnósticos diferenciales. En pacientes con erosiones en la zona de la mucosa oral (“estomatitis aftosa resistente al tratamiento”), posiblemente en combinación con ampollas/erosiones en la piel queratinizada, siempre debe considerarse la presencia de una dermatosis bullosa autoinmune como diagnóstico diferencial. Los antecedentes de epistaxis, disfagia y disnea son esenciales en este caso, al igual que una inspección detallada de la mucosa conjuntival y genital. El tratamiento de las dermatosis ampollosas autoinmunes debe realizarse en centros especializados. Con nuevas opciones terapéuticas como el rituximab y la inmunoadsorción, incluso las formas graves de la enfermedad pueden tratarse bien en la mayoría de los casos. La determinación del antígeno diana no sólo es importante para un diagnóstico exacto, sino que también puede indicar la presencia de un proceso paraneoplásico.
Las dermatosis bullosas autoinmunes son un grupo de enfermedades autoinmunológicas que tienen en común la presencia de autoanticuerpos contra moléculas estructurales y de adhesión de la piel o las mucosas. Como resultado de la desregulación de la inmunidad adaptativa tanto humoral como celular, se desarrollan hendiduras que se asocian clínicamente con ampollas y erosiones en la piel y/o las mucosas.
En cuanto a la localización de la formación de la hendidura, es posible dividir las dermatosis bullosas autoinmunes en dos grandes grupos orientativos:
1. Dermatosis bullosas autoinmunes con pérdida de adherencia intraepitelial
– Enfermedades del pénfigo
2. dermatosis bullosas autoinmunes con pérdida subepitelial de adherencia
– Enfermedades penfigoides
– Dermatosis IgA lineal
– Epidermólisis bullosa adquirida
– Dermatitis herpetiforme Duhring
Enfermedades del pénfigo
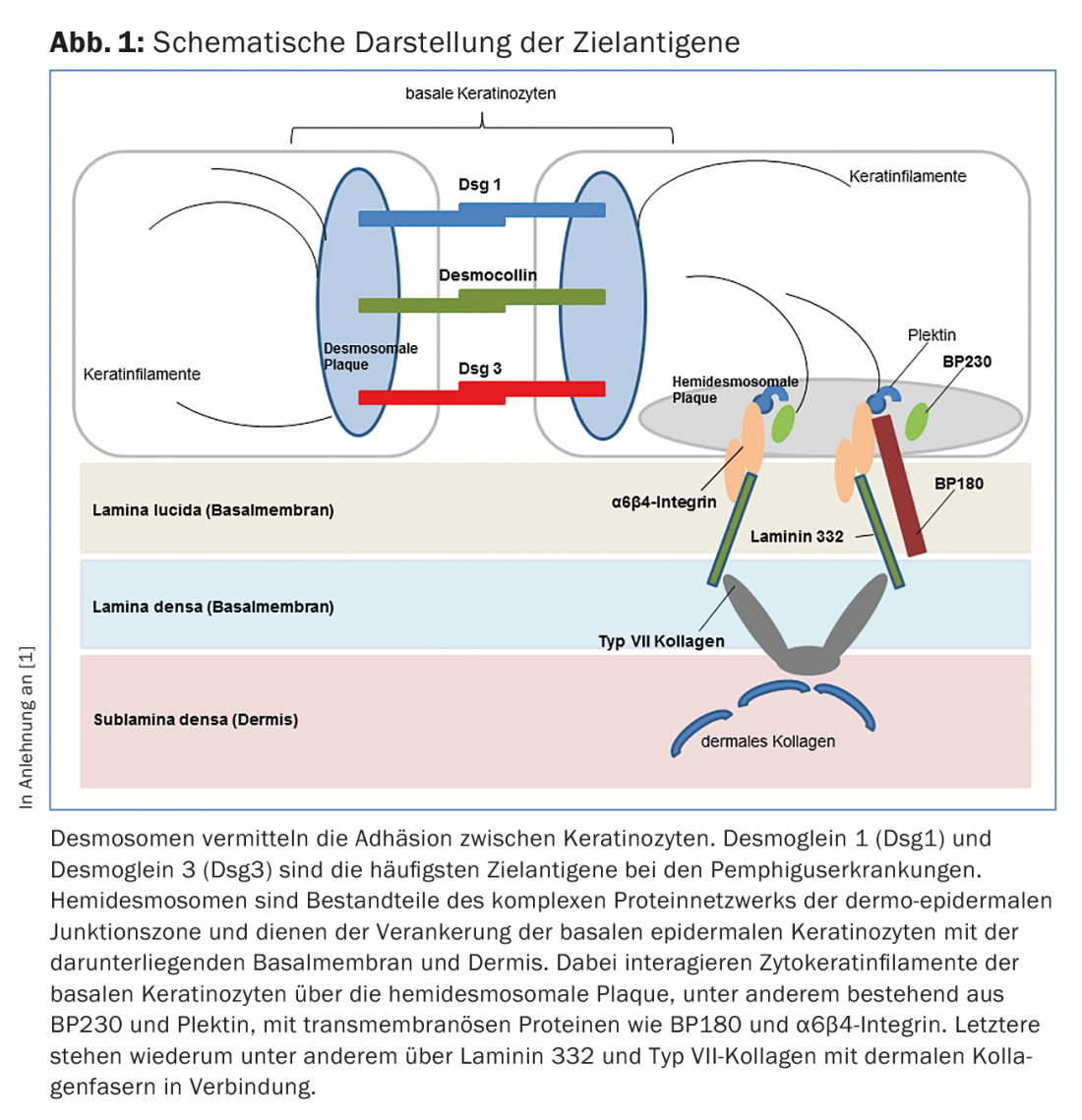
En las enfermedades del grupo del pénfigo, se produce una pérdida de contacto intraepitelial célula-célula debido a autoanticuerpos contra proteínas de los desmosomas (Fig. 1), lo que da lugar a la formación de hendiduras intraepidérmicas localizadas superficialmente. Desde el punto de vista clínico e inmunopatológico, pueden distinguirse diferentes formas de pénfigo.
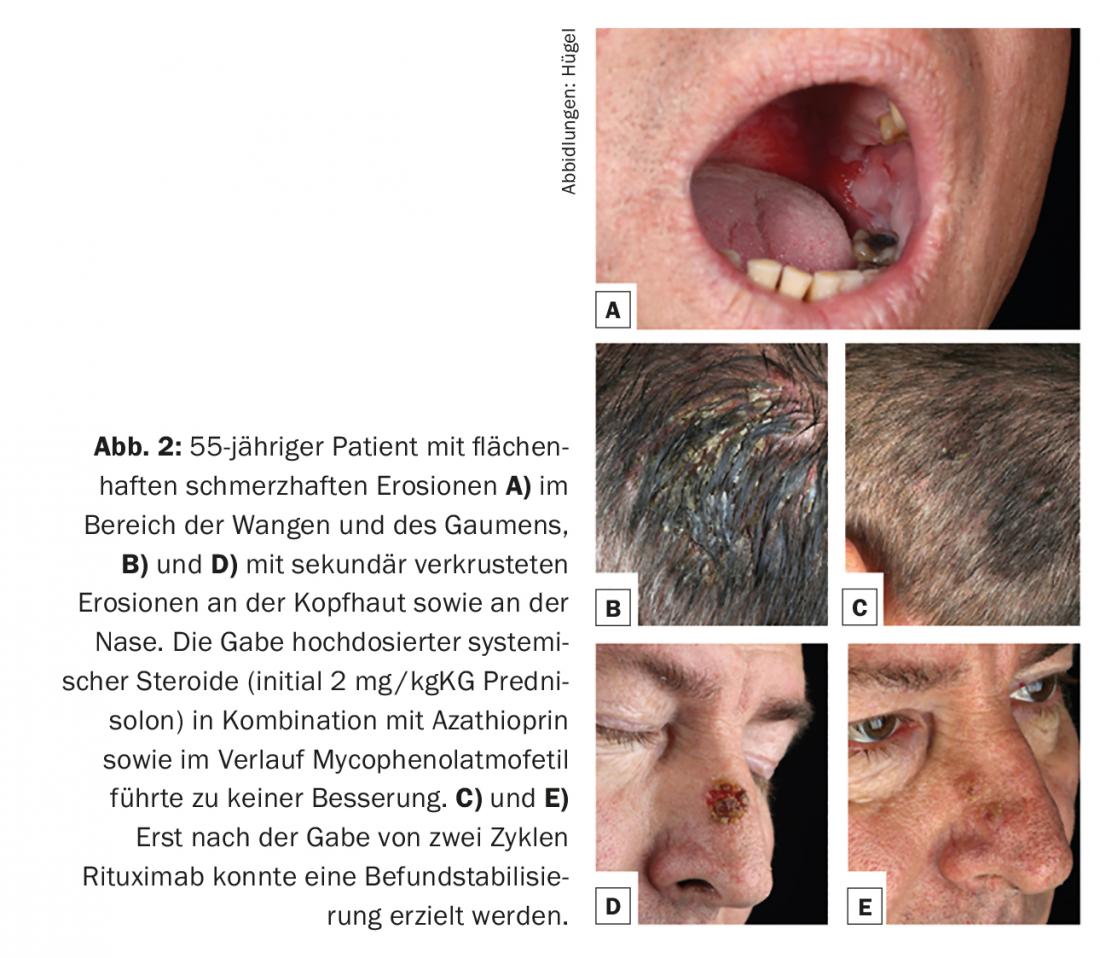
El pénfigo vulgar representa aproximadamente el 80% de los casos de pénfigo, manifestándose preferentemente entre la cuarta y la sexta décadas de la vida, con una incidencia de 0,1-0,5/100.000/año. Desde el punto de vista etiopatogénico, siempre se encuentran autoanticuerpos contra la desmogleína 3. Al principio de la enfermedad, suele haber erosiones enorales de la mucosa muy dolorosas (Fig. 2A) y no es infrecuente que la primera presentación sea ante un especialista en enfermedades otorrinolaringológicas. Opcionalmente, los anticuerpos contra la desmogleína 1 también se encuentran en el pénfigo vulgar, con infestación consecutiva de la piel queratinizada. (Fig. 2B y D). El aspecto clínico de la piel se caracteriza por ampollas extremadamente vulnerables con un fino techo ampolloso, que se rompen rápidamente y, por lo tanto, normalmente ya no se presentan como ampollas, sino como erosiones (secundariamente incrustadas). El fenómeno Nikolski I (empujabilidad de las capas epidérmicas superiores por tracción tangencial sobre la piel intacta) puede inducirse en el examen clínico.
En cambio, en la segunda enfermedad más común del pénfigo -el pénfigo foliáceo- sólo se encuentran autoanticuerpos contra la desmogleína 1, pero no contra la desmogleína 3. De acuerdo con el patrón de expresión de las desmogleínas, esta enfermedad afecta por tanto exclusivamente a las zonas córneas de la piel y no a las mucosas. Aquí tampoco se encuentran casi nunca ampollas intactas, sino erosiones extensas con descamación a veces en forma de hoja. Los síntomas cutáneos suelen comenzar en la cabeza peluda, la cara o la zona de los surcos sudoríparos delanteros y traseros, y luego se extienden a la periferia.
El pénfigo par aneoplásico, que se da en muy raras ocasiones, se asocia en particular a neoplasias hematológicas (especialmente linfomas no Hodgkin de células B) y presenta autoanticuerpos contra dianas desmosómicas y no desmosómicas. Clínicamente, se caracteriza por erosiones dolorosas extensas y úlceras de las membranas mucosas (especialmente boca, labios, esófago), afectación conjuntival y lesiones cutáneas polimorfas.
El pénfigo IgA (caracterizado por depósitos intraepidérmicos de IgA en la biopsia cutánea para inmunofluorescencia directa, Fig. 3C) es la más rara de las variantes de pénfigo. Clínicamente, las pústulas y vesículas sobre una base eritematosa se encuentran principalmente en las zonas intertriginosas (Fig. 3A y B).
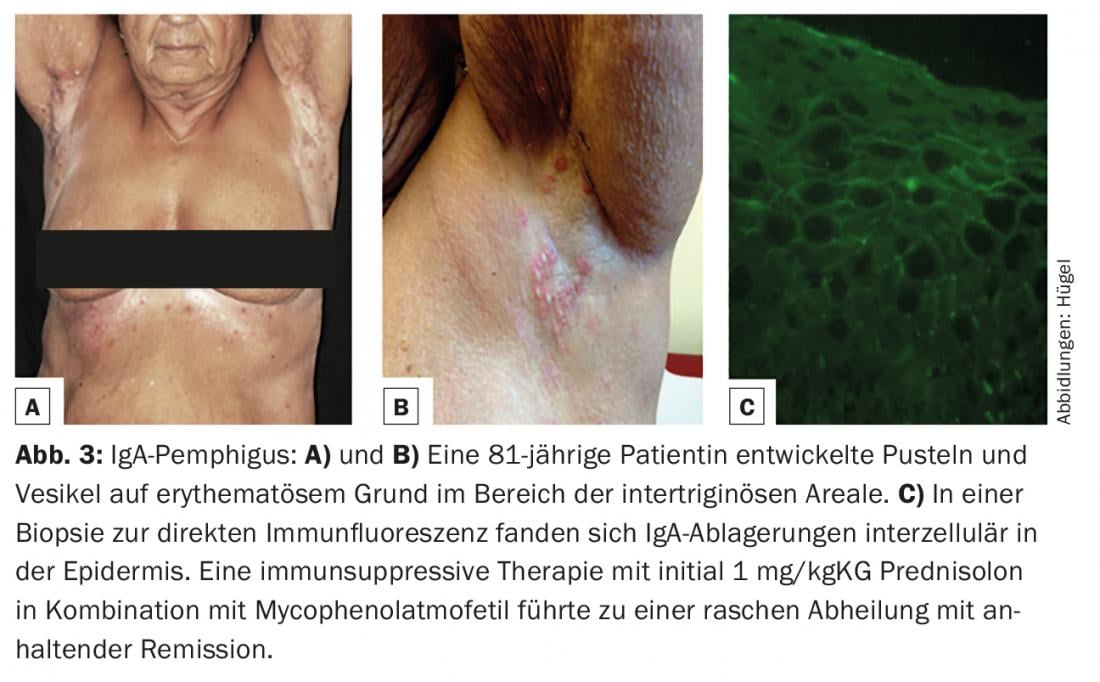
Todos los pacientes diagnosticados de pénfigo deben tener un cuidadoso historial de medicación, ya que fármacos como la penicilamina (que, sin embargo, se utiliza muy raramente en el tratamiento de enfermedades reumáticas en la actualidad), pero también los inhibidores de la ECA como el captopril, el enalapril o el lisinopril pueden inducir la enfermedad del pénfigo.
Dermatosis ampollosas autoinmunes con pérdida subepitelial de adherencia
En las enfermedades penfigoides, las ampollas de la piel parecen mucho más estables y regordetas que en las enfermedades penfigóticas debido a la formación de una hendidura subepidérmica más profunda. Los pacientes suelen sufrir picores intensos. El cuadro clínico es heterogéneo.
El penfigoide bulloso (PB) es la dermatosis autoinmune bullosa más común, con una incidencia de 12-21 casos por 1.000.000 de habitantes/año. La principal edad de aparición se sitúa entre los 60 y los 90 años y, como consecuencia del aumento general de la esperanza de vida, la incidencia ha aumentado considerablemente en los últimos años. Un rasgo característico del penfigoide ampolloso es la presencia de autoanticuerpos contra dos proteínas estructurales hemidesmosómicas de la zona de la membrana basal: BP180 y/o (más raramente) BP230. (Fig.1). Clínicamente, el penfigoide ampolloso se presenta con ampollas abultadas llenas de líquido seroso, eritema diseminado y lesiones urticariales seguidas de erosiones y costras. (Fig. 4A y B). Las mucosas se ven afectadas en un 10-30%. En una fase premonitoria, la enfermedad puede progresar durante meses o años sin que se formen ampollas. Por lo tanto, en los pacientes ancianos con prurito intenso y lesiones cutáneas polimorfas (focos de eccema, placas urticariales, excoriaciones como consecuencia del prurito), debe considerarse siempre como diagnóstico diferencial un estadio prebulloso de la PA.
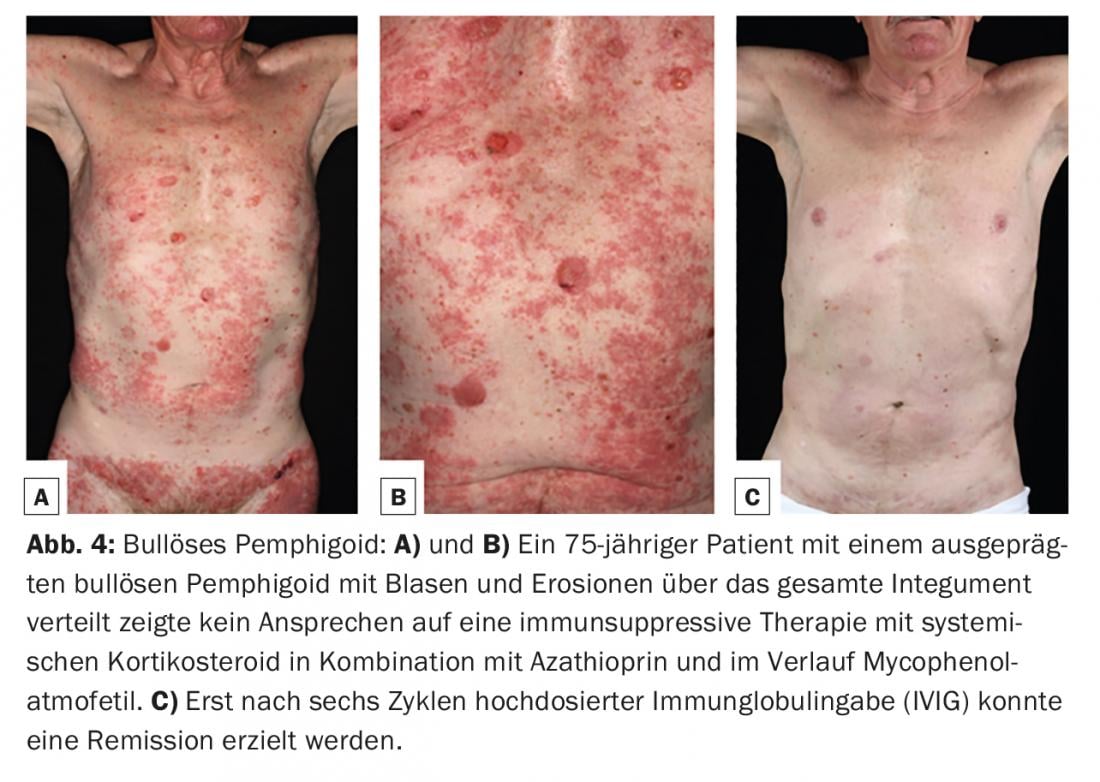
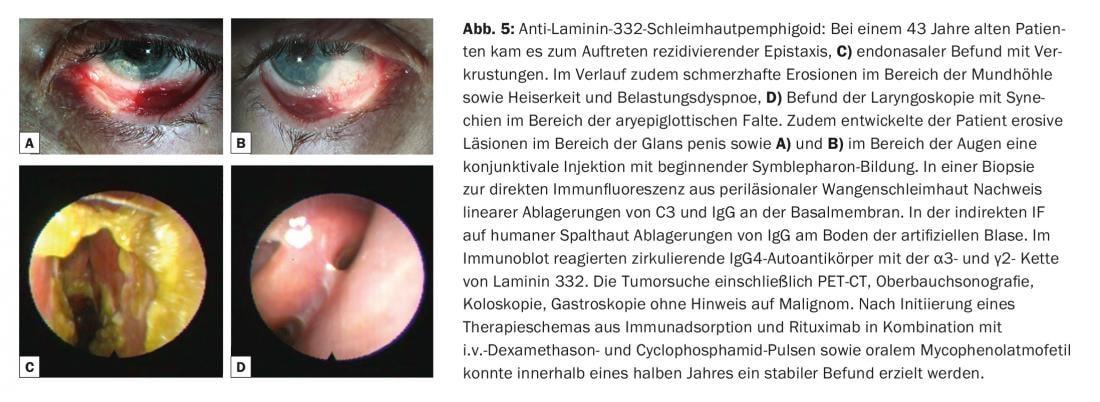
El término penfigoide de las mucosas abarca un grupo heterogéneo de enfermedades raras del espectro de las dermatosis autoinmunes bullosas, que se asocian a la formación de ampollas subepiteliales o subepidérmicas y en las que los cambios inflamatorios crónicos se producen predominantemente en la zona de las mucosas. La formación de ampollas está causada por autoanticuerpos contra las moléculas de adhesión de la zona de unión dermoepidérmica, y se han identificado al menos diez antígenos diana diferentes. Lo más frecuente es encontrar autoanticuerpos circulantes IgG y/o IgA contra el BP180. Con menor frecuencia, se detectan autoanticuerpos contra BP230, laminina-332, α6β4-integrina o colágeno VII (Fig. 1). La identificación del antígeno diana es de gran importancia, ya que se ha descrito una asociación con neoplasias malignas en el 30% de los casos para un subtipo (penfigoide anti-laminina-332) y debe realizarse la exclusión tumoral correspondiente. Clínicamente, todas las mucosas con epitelio escamoso estratificado pueden verse afectadas en el penfigoide mucoso. La afectación más frecuente es la de la mucosa oral, inicialmente con la aparición de una gingivitis descamativa. La afectación ocular suele comenzar unilateralmente con el cuadro clínico de una conjuntivitis, en cuyo curso se produce un acortamiento de los pliegues conjuntivales (fórnices), simblefaron, triquiasis, sinequias y atrofia corneal con un eventual riesgo de ceguera. (Fig. 5A y B). Se encuentran costras endonasales, epistaxis, disfagia, ronquera y estridor con afectación nasofaríngea o laríngea. (Fig. 5C y D). En los hombres, las infestaciones genitales pueden causar adherencias entre el glande del pene y el prepucio, y en las mujeres, obstrucción del introito vaginal. La afectación cutánea – clínicamente similar al penfigoide bulloso – se encuentra en alrededor del 20% de todos los casos.
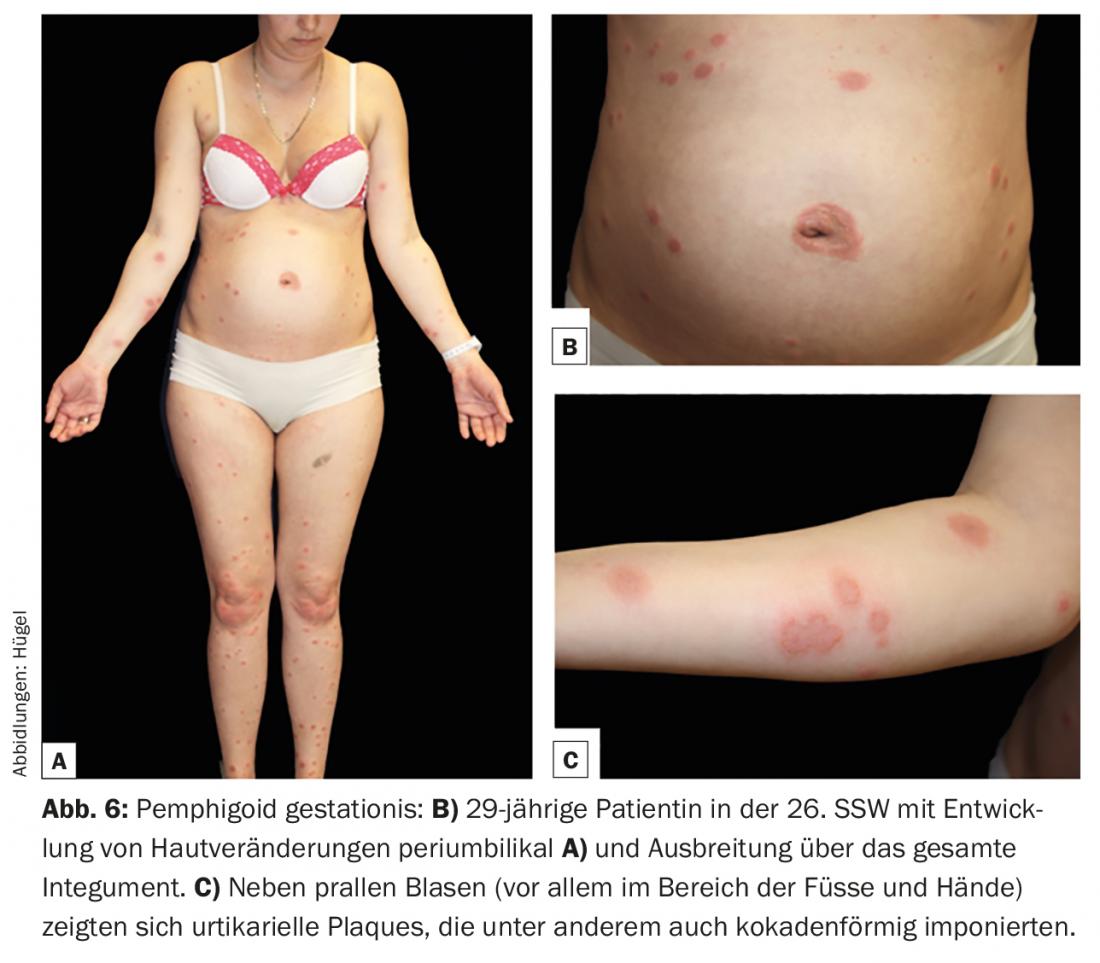
El penfigoide gestacional (PG) es una rara dermatosis del embarazo que puede aparecer en el segundo y tercer trimestres o inmediatamente después del parto y es fisiopatológicamente muy parecida al penfigoide bulloso. Como en este caso, el BP180 y -mucho más raramente- el BP230 son los antígenos diana cruciales. Aún no se ha aclarado la causa del desarrollo de autoanticuerpos. El sistema HLA parece desempeñar un papel importante en la predisposición genética. Se discute que la presentación de autoantígenos placentarios junto con moléculas HLA de clase II paternas conduce a una inducción de autoanticuerpos. Clínicamente, el desarrollo de eflorescencias específicas suele ir precedido de una fase prodrómica con prurito intenso. Las lesiones cutáneas suelen comenzar periumbilicalmente (Fig. 6B) y luego pueden extenderse por todo el tegumento. Las lesiones más comunes son pápulas y/o placas urticariales, que en ocasiones aparecen en forma de coqueta. Sólo en el curso de la enfermedad se desarrollan vesículas agrupadas o ampollas abultadas en la mayoría de los pacientes (Fig. 6).
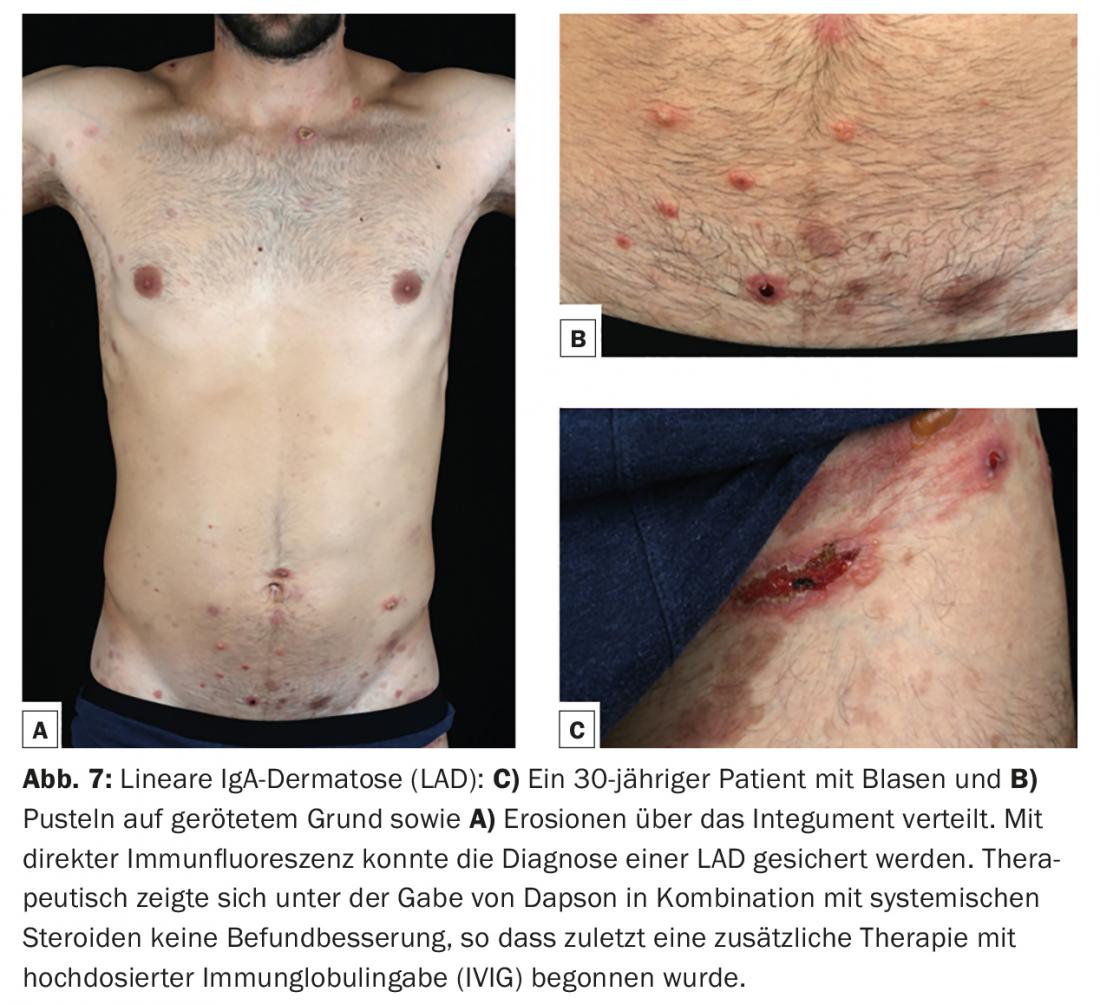
La dermatosis IgA lineal (DAI) es la dermatosis ampollosa autoinmune más común de la infancia y suele comenzar antes de los seis años. En la edad adulta, la manifestación inicial es posible a cualquier edad, pero más a menudo después de los 60 años. Al igual que el penfigoide mucoso, se considera un cuadro clínico heterogéneo. Son característicos los depósitos lineales de tipo IgA a lo largo de la zona de unión dermoepidérmica en la biopsia cutánea para inmunofluorescencia directa. Los autoanticuerpos IgA se dirigen más comúnmente contra una proteína de 97 kDa (LABD-97) y una proteína de 120 kDa (LAD-1), que se producen por escisión proteolítica de la parte extracelular de la BP180. Clínicamente, suelen aparecer ampollas anulares o policíclicas sobre piel sana o eritematosa con afectación preferente de la cara (especialmente perioral y orejas), región anogenital y, con menor frecuencia, tronco, manos y pies. (Fig.7). Pueden producirse cicatrices en las membranas mucosas, especialmente en la conjuntiva, como en el penfigoide de las membranas mucosas, e incluso provocar ceguera.
El cuadro clínico de la epidermólisis ampollosa adquirida (EBA), en la que etiopatogénicamente están presentes anticuerpos de clase IgG (e IgA) contra el colágeno VII (Fig. 1) , es muy variable. En la variante mecano-bullosa, la piel es muy vulnerable y se encuentran ampollas y erosiones que curan cicatrizadas o con milia, especialmente en zonas sometidas a esfuerzos mecánicos como el dorso de la mano, el codo, la rodilla, la región sacra y los dedos de los pies. En las variantes inflamatorias, esta enfermedad muestra un cuadro clínico que puede ser difícil de distinguir del penfigoide ampolloso o del penfigoide mucoso.
La dermatitis herpetiforme Duhring es un subtipo cutáneo poco frecuente de las enfermedades sensibles al gluten. Hace unos años, la transglutaminasa epidérmica (transglutaminasa 3) y transglutaminasa tisular (transglutaminasa 2). Los pacientes con esta enfermedad desarrollan pápulas y vesículas principalmente en los lados extensores de las extremidades, en la cabeza vellosa, glútea y sacra. Se trata de una afección que provoca mucho picor. Por lo tanto, debe considerarse la enfermedad de Duhring en todo paciente con excoriaciones por rascado en primer plano en la zona de los lugares de predilección mencionados.
Diagnóstico de las dermatosis bullosas autoinmunes
Las biopsias cutáneas para un examen histológico convencional pueden utilizarse para mostrar la localización de la hendidura.
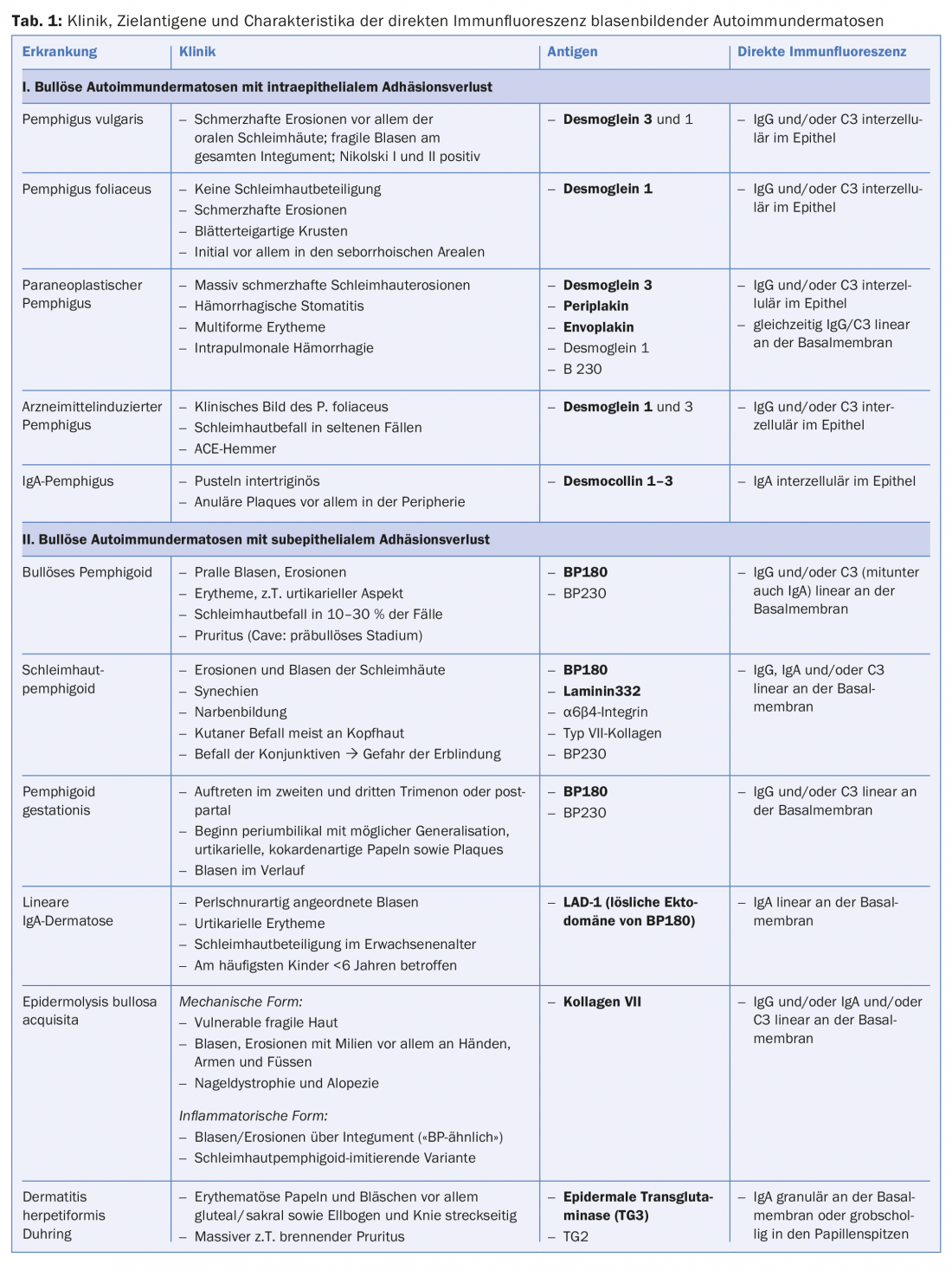
Sin embargo, la regla de oro diagnóstica es la detección de autoanticuerpos unidos a tejidos (IgG, IgA y/o factor del complemento C3) en biopsias de piel o mucosas con inmunofluorescencia directa. La localización de los anticuerpos intercelularmente en la epidermis o a lo largo de la zona de unión dermoepidérmica permite diferenciar inmediatamente entre la presencia de una enfermedad pénfigo o una enfermedad del espectro de las dermatosis bullosas autoinmunes subepiteliales. Si predominan los precipitados del isotipo IgA, puede diagnosticarse un pénfigo IgA (fig. 3C), una dermatosis IgA lineal o una enfermedad de Duhring, según el patrón (tab. 1).
Además de las biopsias cutáneas (mucosas) y para la caracterización exacta de los distintos subtipos, se requieren exámenes serológicos. La inmunofluorescencia indirecta en esófago de mono y en piel salina humana se ha establecido como prueba de detección para este fin. A continuación, se identifican los antígenos diana con ayuda de diversos ELISA y, si es necesario, de exámenes inmunoblot específicos.
Las concentraciones de autoanticuerpos en el suero de los pacientes con pénfigo y penfigoide suelen correlacionarse bien con la actividad de la enfermedad y, por lo tanto, también son adecuadas para controlar la actividad de la enfermedad y evaluar la necesidad de una terapia adicional.
Terapia de las dermatosis bullosas autoinmunes
En general, la terapia de las dermatosis ampollosas autoinmunes depende de la gravedad, por un lado, y del subtipo diagnosticado, por otro. En el caso, por ejemplo, de un penfigoide ampolloso localizado leve, puede ser suficiente el tratamiento local únicamente con esteroides de clase IV muy potentes (pomada de propionato de clobetasol al 0,05%). En la mayoría de los casos, sin embargo, está indicada la administración sistémica de corticosteroides en combinación con otros inmunosupresores (por ejemplo, azatioprina, micofenolato mofetilo, ciclosporina, metotrexato, ciclofosfamida). La dapsona es el agente de primera línea para la dermatosis IgA lineal, la dermatitis herpetiforme Duhring y el penfigoide mucoso oral puro no complicado. En casos graves o refractarios, pueden utilizarse inmunoglobulinas intravenosas (IGIV), inmunoadsorción y/o rituximab. Además, en los últimos años se han publicado varios informes de casos que demuestran la eficacia de la terapia con el anticuerpo anti-IgE omalizumab en el penfigoide ampolloso. Sin embargo, esto requiere más estudios de normalización en lo que respecta a la dosis, así como a la duración del uso.
La atención a los pacientes con dermatosis ampollosas autoinmunes debe realizarse en centros especializados y requiere -especialmente en presencia de un subtipo con afectación de las mucosas- un enfoque interdisciplinar con la participación de oftalmólogos, otorrinolaringólogos, dentistas, ginecólogos, gastroenterólogos y el especialista dermatológico como coordinador.
Literatura:
- Schmidt E, Zillikens E: Diagnóstico y terapia de las dermatosis bullosas autoinmunes. Dtsch Arztebl Internacional 2011; 108(23): 399-405.
Para saber más:
- Hertl M (ed.): Enfermedades autoinmunes de la piel. Patogénesis, diagnóstico, gestión.3ª edición. Viena – Nueva York: Springer-Verlag 2011.
- Marazza G, et al: Incidencia del penfigoide bulloso y el pénfigo en Suiza: un estudio prospectivo de 2 años. Br J Dermatol 2009; 161(4): 861-868.
- Kneisel A, Hertl M: Enfermedades ampollosas autoinmunes de la piel. Parte 1: Manifestaciones clínicas. J Dtsch Dermatol Ges 2011; 9(10): 844-856.
- Kneisel A, Hertl M: Enfermedades ampollosas autoinmunes de la piel. Parte 2: diagnóstico y terapia. J Dtsch Dermatol Ges 2011; 9(11): 927-947.
- Kneisel A, Hertl M: Penfigoide bulloso: diagnóstico y terapia. Wien Med Wochenschr 2014; 164(17-18): 363-371.
- Schmidt E, Zillikens D: Enfermedades penfigoides. Lancet 2013; 381(9863): 320-332.
- Hertl M, et al.: Recomendaciones para el uso de rituximab (anticuerpo anti-CD20) en el tratamiento de enfermedades autoinmunes ampollosas de la piel. J Dtsch Dermatol Ges 2008; 6(5): 366-373.
PRÁCTICA DERMATOLÓGICA 2017; 27(1): 18-25