
La sarcoidosis es una enfermedad granulomatosa multisistémica de etiología aún poco clara. El diagnóstico de sarcoidosis se basa en el cuadro clínico típico y la evidencia histológica de granulomas de células epitelioides no necrotizantes, tras la exclusión de otros diagnósticos diferenciales. Dado que más del 90% de los pacientes con sarcoidosis tienen un pulmón o una inflamación pulmonar, es importante que los pulmones no resulten dañados. Si también están afectados los ganglios linfáticos intratorácicos, el diagnóstico suele realizarse mediante biopsias obtenidas por broncoscopia. Si está indicado, la terapia se lleva a cabo con inmunosupresores o fármacos inmunosupresores. Inmunomoduladores.
La sarcoidosis es una enfermedad granulomatosa multisistémica de etiología aún poco clara [1]. Se caracteriza por granulomas de células epitelioides, no necrotizantes, en diversos órganos, por lo que tiene una clínica variable según el órgano afectado. No existe ninguna prueba diagnóstica específica, por lo que el diagnóstico se basa en los hallazgos clínicos típicos, la detección de los granulomas típicos y la exclusión de los diagnósticos diferenciales.
La prevalencia de la sarcoidosis se estima entre 1 y 40/100.000 habitantes en todo el mundo, aunque las diferentes definiciones de los casos, la presentación clínica variable y, en última instancia, la dificultad ocasional para realizar un diagnóstico dificultan una cuantificación precisa [2,3]. Además, se ha descrito una “división Norte-Sur” con una mayor prevalencia en Escandinavia [4]; el estudio ACCESS confirmó que los afroamericanos padecen la enfermedad con mayor frecuencia y gravedad [5].
En Suiza, se encontró una prevalencia a lo largo de la vida de 121/100.000 para el diagnóstico de sarcoidosis (130/100.000 hombres, 112/100.000 mujeres). Se calculó una prevalencia de 44/100.000 para la sarcoidosis activa y de 16/100.000 para la sarcoidosis que requirió hospitalización. La incidencia media anual en Suiza se estima en 7/100.000 habitantes [6].
La enfermedad es posible a cualquier edad, aunque los adultos más jóvenes (<40 años) tienen más probabilidades de desarrollar la enfermedad; existe un segundo pico de enfermedad para el sexo femenino a la edad de >50 años [7]. En Suiza, la edad media en el momento del diagnóstico es de 45 +/- 15 años (41 +/- 14 años para los hombres, 48 +/- 15 años para las mujeres) [6].
Causa y patogénesis
Se desconocen las causas de la sarcoidosis. En la literatura se discuten varias hipótesis. La enfermedad se desencadena probablemente por la inhalación de un agente/antígeno (patógeno, aerosol) en pacientes genéticamente predispuestos, lo que se ve respaldado por la observación de una incidencia agrupada de sarcoidosis en el primer año tras el atentado terrorista del 11 de septiembre contra el World Trade Center de Nueva York entre los bomberos expuestos al polvo [8]. En Suiza, se observó una mayor prevalencia en las regiones con industrias de transformación de metales y con agricultura (transformación de patatas y cereales y cultivo de prados) [6]. Debido a la característica de una enfermedad granulomatosa, también se habló de exposición a micobacterias u otros patógenos, pero nunca se demostró.
La observación de una acumulación tanto familiar como étnica sugiere que existe un trasfondo genético [9]. Se han estudiado diferentes alelos -principalmente del complejo mayor de histocompatibilidad (CMH) y del gen ACE-, identificando genotipos asociados con mayor frecuencia a la sarcoidosis [10]. Algunos genotipos, a su vez, se han asociado al síndrome de Löfgren y a un curso benigno, entre otros [11].
Los procesos inmunológicos están implicados en la patogénesis de la sarcoidosis, aunque aún no se conocen en detalle. La respuesta inmunitaria se desencadena presumiblemente por la presentación de un antígeno indefinido por los macrófagos a los linfocitos T CD4+ a través de receptores CMH de clase II. Las quimiocinas y citocinas (incluyendo TNF-α, IL-2) liberadas en el proceso conducen a la formación de los típicos granulomas compuestos por células epitelioides, células gigantes multinucleadas y linfocitos T CD4+ [12–14]. Se desconocen los mecanismos que provocan que la enfermedad sea autolimitada por un lado y crónicamente progresiva por otro.
Diagnóstico
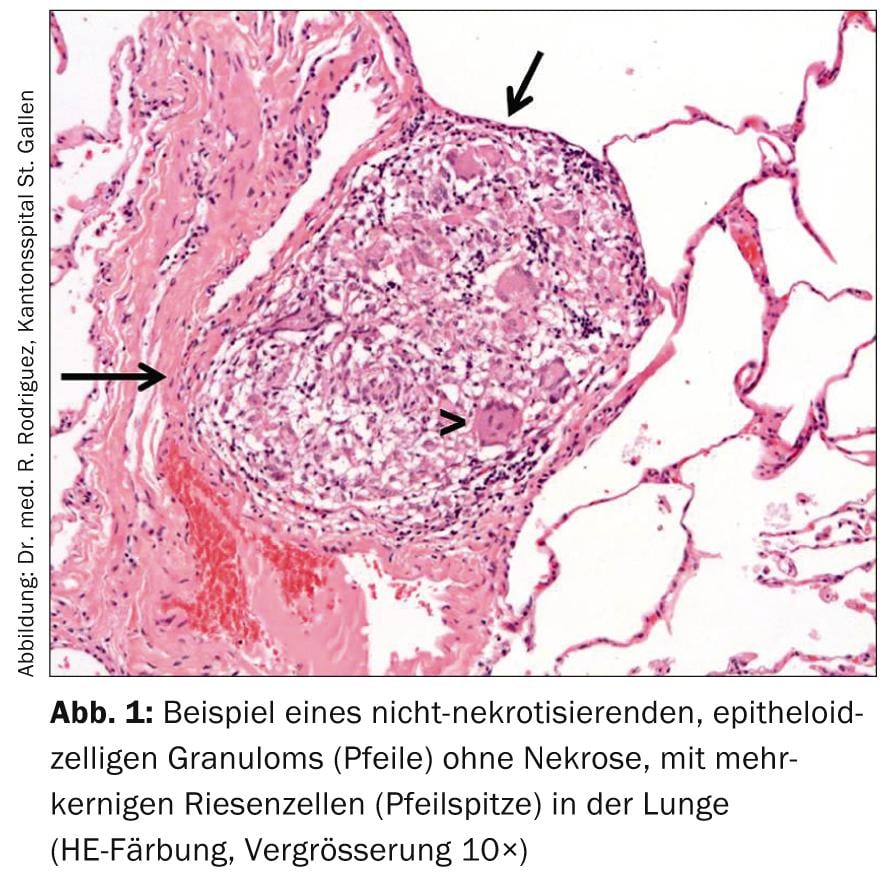
El diagnóstico de sarcoidosis se realiza con un cuadro clínico típico, evidencia histopatológica de granulomas de células epitelioides no necrotizantes (Fig. 1) y tras la exclusión de diagnósticos diferenciales.
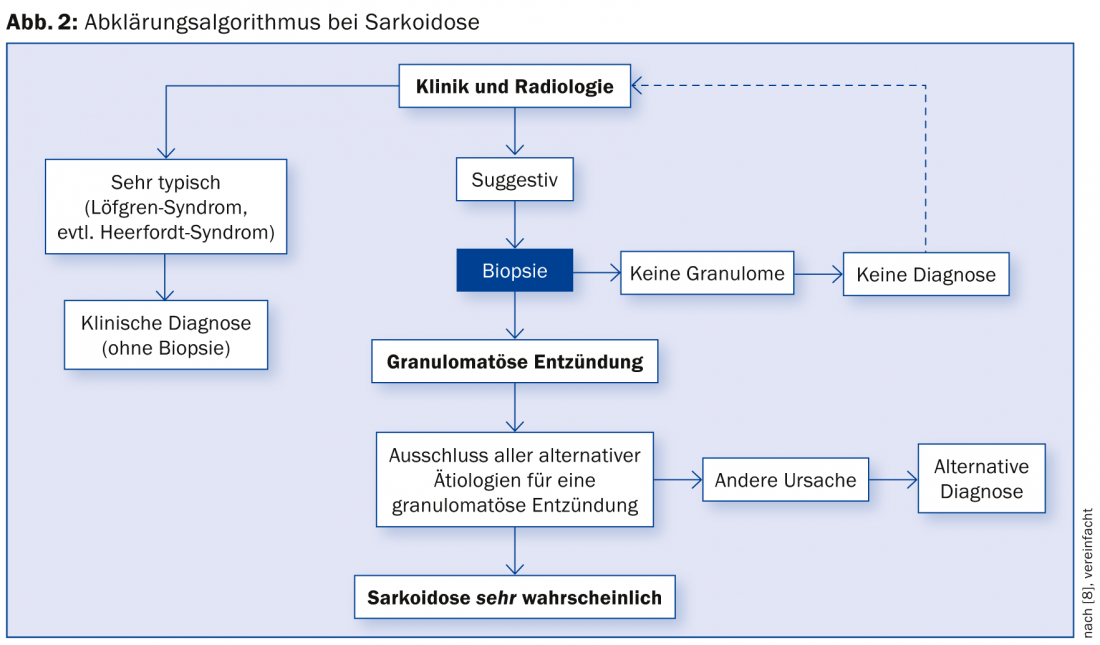
Un posible algoritmo de diagnóstico se resume en la figura 2 [15].
Síntomas típicos
Los granulomas típicos pueden desarrollarse en cualquier órgano. Con frecuencia se ven afectados los ganglios linfáticos hiliares y mediastínicos, el tejido pulmonar, la piel y las estaciones ganglionares extratorácicas. Con menor frecuencia, se describe afectación oftalmológica, neurológica o cardiaca (tab. 1); cuando estos órganos se ven afectados, el deterioro clínico, la morbilidad y la mortalidad son significativos [16]. No existe una única sintomatología típica de la sarcoidosis, ya que prácticamente todos los órganos pueden verse afectados.

Los pacientes suelen ser asintomáticos, por lo que no es infrecuente que la sarcoidosis sea un hallazgo incidental en una linfadenopatía bihiliar cuando se realiza una radiografía convencional [17].
Dado que las manifestaciones pulmonares están presentes en más del 90% de los pacientes, son comunes síntomas como la tos no productiva, la disnea y el dolor torácico. También suelen describirse síntomas generales inespecíficos como fiebre, pérdida de peso, fatiga crónica, malestar, debilidad muscular e intolerancia al ejercicio.
Síndrome de Löfgren y síndrome de Heerfordt
La combinación de linfadenopatía biliar, fiebre, eritema nodoso y artralgias/artritis se denomina síndrome de Löfgren. Esta clínica es característica, por lo que la evidencia histológica de granulomas puede omitirse en esta situación. El síndrome de Löfgren tiene un buen pronóstico. La rara pero específica combinación de parotitis con uveítis y paresia facial periférica (“parálisis de Bells”) con granulomas sarcoides histológicamente probados se conoce como síndrome de Heerfordt.
Manifestación intratorácica
Las manifestaciones intratorácicas van desde la linfadenopatía bihiliar aislada hasta la destrucción grave del parénquima pulmonar y la hipertensión pulmonar (incluso sin cambios pulmonares). La gravedad pulmonar se clasifica en cinco estadios radiológicos según Siltzbach y Scadding sobre la base de la radiografía convencional (Tab. 2 y Fig. 3) [18,19].

La estadificación es importante porque de ella puede derivarse información pronóstica. Dos tercios de los pacientes experimentan una remisión espontánea en un plazo de 2 a 5 años. Esto es especialmente cierto para los estadios radiológicos I y II en el momento del diagnóstico.
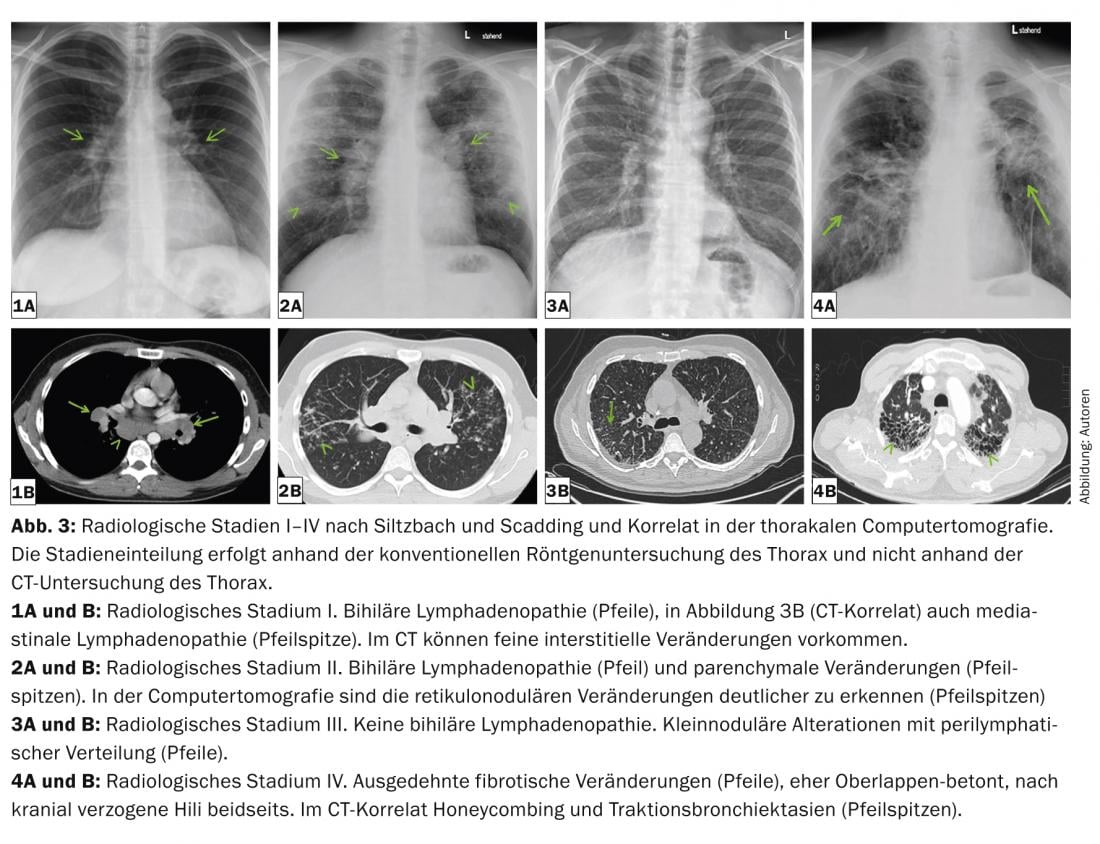
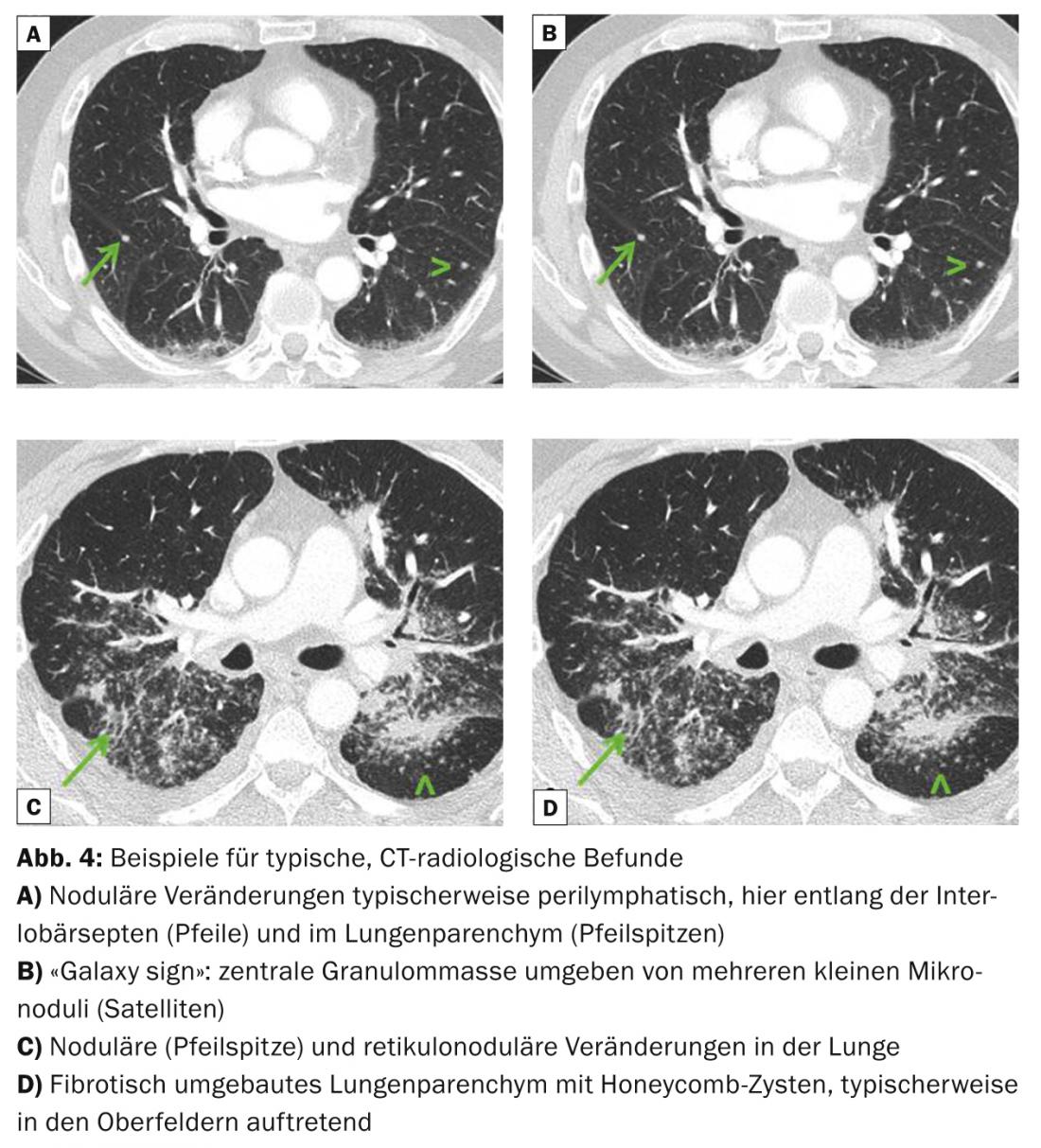
Las imágenes radiológicas pulmonares típicas son una linfadenopatía mediastínica bihiliar, pero también simétrica, micronódulos (diámetro <3 mm) con distribución típica (campos superior y medio, a lo largo de los haces broncovasculares, subpleural y a lo largo de los septos interlobulares), macronódulos bilaterales que raramente pueden fundirse (como expresión de necrosis isquémica del núcleo del granuloma), opacidades en vidrio deslustrado, proliferación de dibujo reticular, fibrosis con panal de abeja, así como bronquiectasias por tracción con énfasis en los campos superiores. (Fig. 3 y 4). La tomografía computarizada de alta resolución (TCAR) es más sensible para evaluar cambios discretos en el parénquima pulmonar.
Manifestación neurológica, cardiaca u oftalmológica
En la sarcoidosis cardiaca suele haber alteraciones de la conducción en el sentido de un bloqueo AV o alteraciones de la conducción intraventricular en el ECG. Los pacientes pueden ser asintomáticos, experimentar palpitaciones o tener síncope o disnea. sufrir arritmias hasta llegar a la muerte súbita cardiaca. Si el miocardio está gravemente afectado, puede haber una cardiomiopatía con el cuadro clínico de una insuficiencia cardiaca. También puede producirse pericarditis.
La afectación oftalmológica suele manifestarse como uveítis, neuritis del nervio óptico e inflamación de las glándulas lagrimales.
En caso de afectación neurológica, lo más frecuente es la neuropatía craneal (paresia facial) o periférica, la irritación meníngea con evidencia de linfocitos en el líquido cefalorraquídeo o la infiltración de la hipófisis o la glándula pituitaria. de la región del hipotálamo con las correspondientes complicaciones endocrinológicas.
La aparición de hipercalcemia puede ser amenazadora, como resultado del aumento de la hidroxilación de la vitamina D3 en las células epiteloides de los granulomas.
Manifestación cutánea
A menudo se describen diversas manifestaciones cutáneas. Son típicos el lupus pernio resp. Eritema nodoso, pero también cambios maculopapulares y nodulares (Fig. 5) – con frecuencia también en la zona de tatuajes y cicatrices. Dependiendo de la localización (cara), las alteraciones cutáneas pueden provocar un importante deterioro subjetivo debido a la desfiguración.
Evidencia histológica de granulomas sarcoides
Excepto en el síndrome de Löfgren, se recomienda la evidencia histológica de granulomas típicos para el diagnóstico; éstos son no necrotizantes, pequeños, fuertemente circunscritos con cierto énfasis perivascular y tendencia a la agregación, y posiblemente fibrosis centrípeta laminar fina. (Fig. 2). La elección del lugar de la biopsia debe permitir el procedimiento menos invasivo posible (por ejemplo, biopsia de piel, biopsia de glándula lagrimal, escisión de ganglios linfáticos periféricos).
Sólo alrededor del 2% de todos los pacientes con sarcoidosis extrapulmonar no presentan afectación pulmonar [16]. Por esta razón, se utilizan técnicas broncoscópicas para tratar de identificar histológicamente o para tomar muestras citológicas. En las lesiones endobronquiales (mucosa alterada de aspecto empedrado en hasta el 50% de los pacientes [20]), puede obtenerse una biopsia de la mucosa mediante biopsia con pinzas; cabe esperar un resultado positivo en más del 60% de los pacientes [21]. Las biopsias pulmonares transbronquiales pueden utilizarse para obtener muestras útiles desde el punto de vista diagnóstico en el 60-97% de los pacientes (dependiendo del número de biopsias tomadas y de la extensión de los cambios radiológicos del parénquima pulmonar) [22–24], y la aspiración transbronquial con aguja (TBNA) guiada por ecografía de los ganglios linfáticos mediastínicos y/o hiliares en aproximadamente el 80% de los pacientes [25]. La combinación de estas técnicas mejora el rendimiento diagnóstico, de modo que la mediastinoscopia, que solía ser necesaria con frecuencia, ahora sólo se utiliza en muy raras ocasiones.
La broncoscopia suele incluir un lavado broncoalveolar. La sarcoidosis es probable -pero no está demostrada- si existen pruebas de alveolitis linfocítica (>15% linfocitos), un cociente CD4/CD8 aumentado >3,5 y se excluye una infección (incluidas las micobacterias). La sensibilidad del cociente CD4/CD8 es del 42-59% y la especificidad del 76-96% para el aumento del cociente >3,5 [26,27].
Mediante la tecnología PET/TC, los focos inflamatorios pueden representarse como regiones con una mayor captación de FDG en casos seleccionados. Si el lugar de la biopsia no puede definirse claramente debido a los hallazgos clínicos, puede utilizarse la tecnología PET/TC para intentar identificar un foco inflamatorio.
Diagnósticos diferenciales a excluir
El diagnóstico de sarcoidosis con un cuadro clínico típico y la evidencia de los hallazgos histopatológicos apropiados puede realizarse cuando se han excluido otras enfermedades granulomatosas. El diagnóstico diferencial incluye enfermedades infecciosas como la tuberculosis, infecciones fúngicas (histoplasmosis, coccidioidomicosis), brucelosis o tularemia. Estas enfermedades pueden descartarse mediante un lavado broncoalveolar si existe una sospecha clínica. Los diagnósticos diferenciales también incluyen enfermedades malignas, especialmente linfomas y carcinomas (exclusión por hallazgos histopatológicos). La beriliosis crónica puede causar un cuadro pulmonar muy similar a la sarcoidosis. Para diferenciar la beriliosis, es importante realizar un historial ocupacional preciso. Si se sospecha, el diagnóstico se realiza mediante la estimulación in vitro de células mononucleares de la sangre o del líquido del lavado broncoalveolar. Además, deben excluirse la alveolitis alérgica exógena (antecedentes, cociente CD4/CD8 disminuido), la neumonitis inducida por fármacos (antecedentes) y los granulomas en el contexto de una reacción a un cuerpo extraño (histología). Dado que los granulomas de la sarcoidosis también pueden aparecer perivascularmente, la diferenciación de una vasculitis en el material de la biopsia puede ser difícil.
Los granulomas pequeños y bien definidos en los ganglios linfáticos de drenaje, en el estroma circundante, pero también en el hígado o el bazo de pacientes con enfermedades malignas se denominan “reacción de tipo sarcoide”; dichos granulomas se producen en aproximadamente el 4% de los pacientes con carcinomas (incluido el cáncer de mama, así como los carcinomas renales y gastrointestinales) y algo más frecuentemente en pacientes con neoplasias malignas de Hodgkin (14%) y no Hodgkin (7%). [28].
Otro diagnóstico diferencial importante cuando se detecta una enfermedad granulomatosa es la inmunodeficiencia común variable (IDCV). Se caracteriza por niveles bajos de inmunoglobulina sérica, infecciones bacterianas recurrentes y una respuesta reducida de anticuerpos. Algunos de los pacientes desarrollan una inflamación granulomatosa con granulomas no necrotizantes en los pulmones, el bazo, el hígado y los ganglios linfáticos, entre otros [29].
La sarcoidosis se trata, entre otras cosas, con inhibidores del TNF-α. Paradójicamente, se ha descrito inflamación granulomatosa durante la terapia con inhibidores del TNF-α, especialmente etanercept en la artritis reumatoide, pero también infliximab [30]. Los granulomas no calcificantes también pueden aparecer en relación con la terapia con interferón-γ (por ejemplo, en la hepatitis C crónica) [31].
Trabajo y evaluación del progreso
Una vez realizado el diagnóstico de sarcoidosis, se recomienda llevar a cabo un estudio de la extensión de la enfermedad. En particular, deben examinarse las manifestaciones orgánicas que puedan provocar una morbilidad importante (Tab. 3).
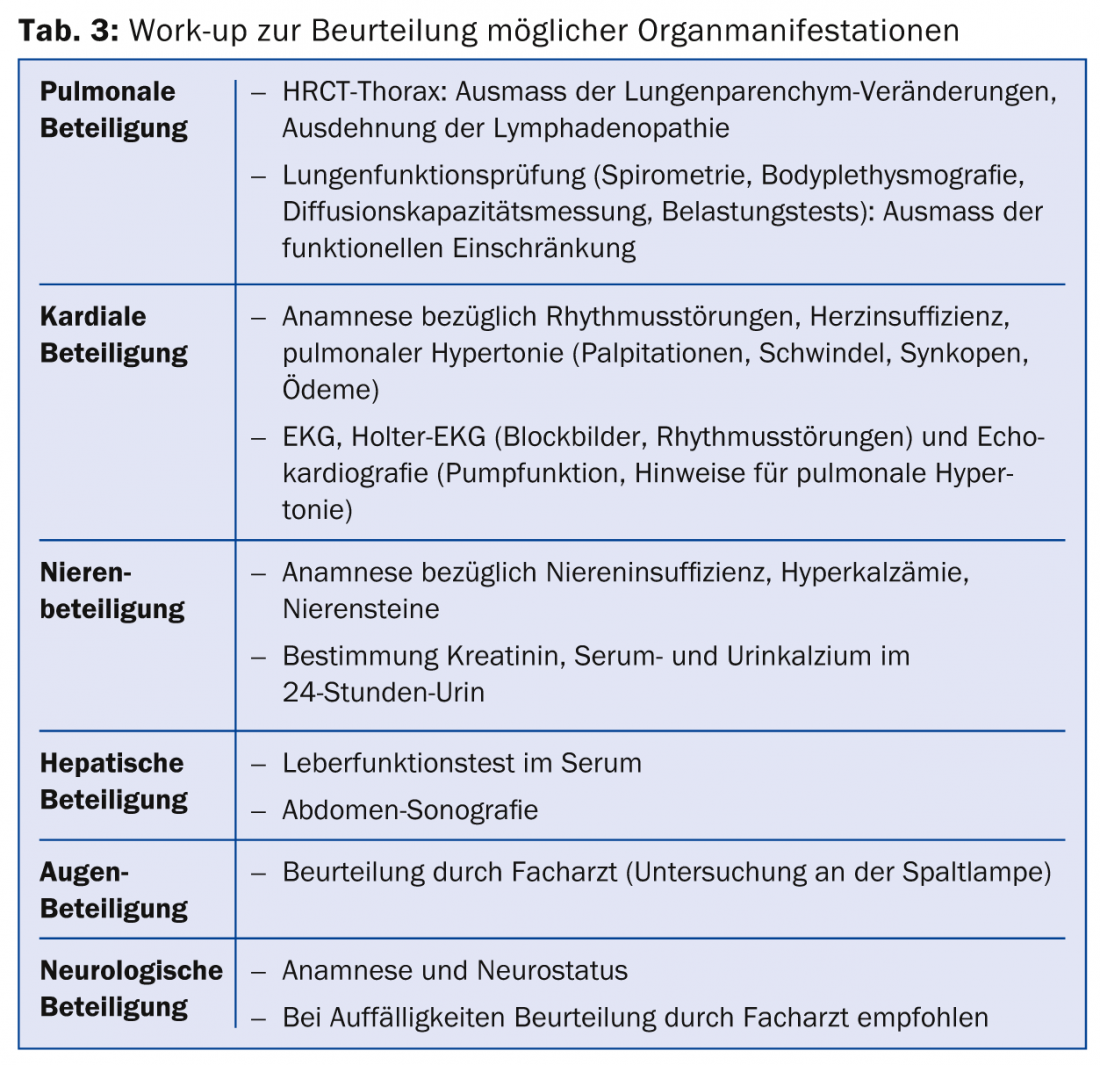
Además, debe definirse qué parámetros pueden utilizarse para la evaluación del curso (por ejemplo, también bajo terapia). Se determina la enzima convertidora de angiotensina (ECA) para evaluar una posible afectación medular. La ECA se produce, entre otras cosas, en las células epiteloides de los granulomas de la sarcoidosis [32]. Sin embargo, debido a su baja sensibilidad, la ACE no es adecuada como prueba diagnóstica. Si está elevada en el momento del diagnóstico, la ECA es un posible parámetro de seguimiento. Otro posible parámetro de la progresión es el receptor soluble de interleucina-2: las células presentadoras de antígenos producen interleucina 2 (IL-2) en relación con la formación de granulomas. Esto conduce a la activación de las células T. Esto libera una forma soluble del receptor de la interleucina-2 (receptor sIL-2) en el torrente sanguíneo. Dado que un aumento del receptor sIL-2 refleja la activación inespecífica de los linfocitos T, con él se puede determinar la actividad de la enfermedad y, por tanto, evaluar el curso.
Pronóstico e indicación de terapia
No se puede predecir el curso de la enfermedad. Sin embargo, el pronóstico es favorable en cerca de dos tercios de los pacientes: la remisión se produce en una década y en cerca del 50% de los pacientes en tres años. En aproximadamente un tercio de los pacientes, la enfermedad progresa de forma crónica con un deterioro funcional relevante de los órganos afectados. Menos del 5% de todos los pacientes mueren de sarcoidosis. La mortalidad se debe principalmente a la fibrosis pulmonar con insuficiencia respiratoria, afectación cardiaca o neurológica o hipertensión pulmonar [33].
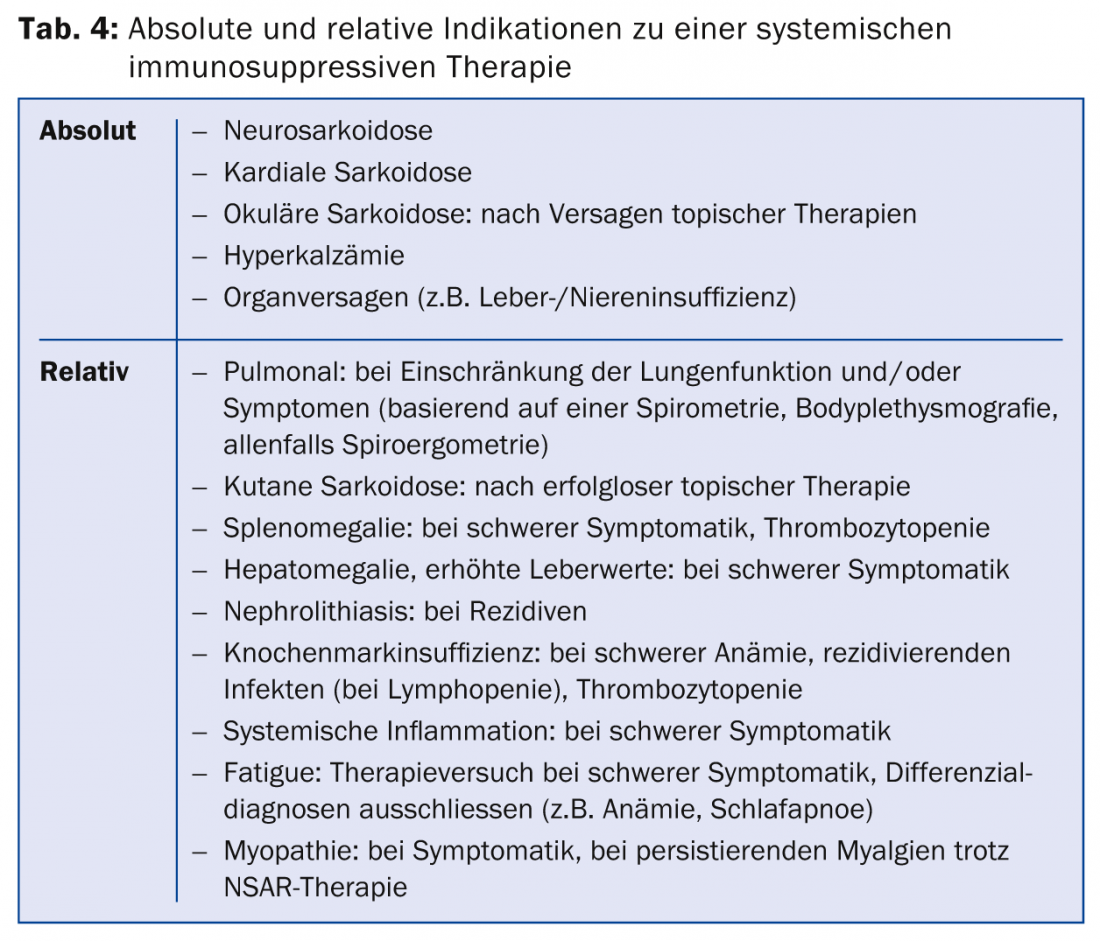
La indicación de la terapia debe hacerse con mucho cuidado, a sabiendas de la evolución favorable de la enfermedad, en su mayor parte espontánea, de los posibles efectos secundarios de la terapia y del aumento de la tasa de recidiva tras la terapia con inmunosupresores (el antígeno postulado persiste en el órgano afectado, por lo que la formación de granulomas puede reaparecer tras la interrupción de la terapia). Existe una indicación absoluta de terapia en casos de neurosarcoidosis, sarcoidosis cardiaca u ocular con afectación del segmento medio o posterior del ojo, así como en casos de deterioro funcional grave de los órganos afectados, típicamente insuficiencia hepática o renal, pero también disfunción pulmonar grave. En el caso de la limitación leve y moderada, es posible una observación clínica cuidadosa del curso: si los síntomas están presentes y la limitación funcional es progresiva, y por lo tanto hay indicios de actividad de la enfermedad, se suele iniciar la terapia.
Terapia con inmunosupresores/inmunomoduladores
Si existe una indicación de terapia (tab. 4), se utilizan inmunosupresores o fármacos inmunosupresores. se utilizan inmunomoduladores (tab. 5) .
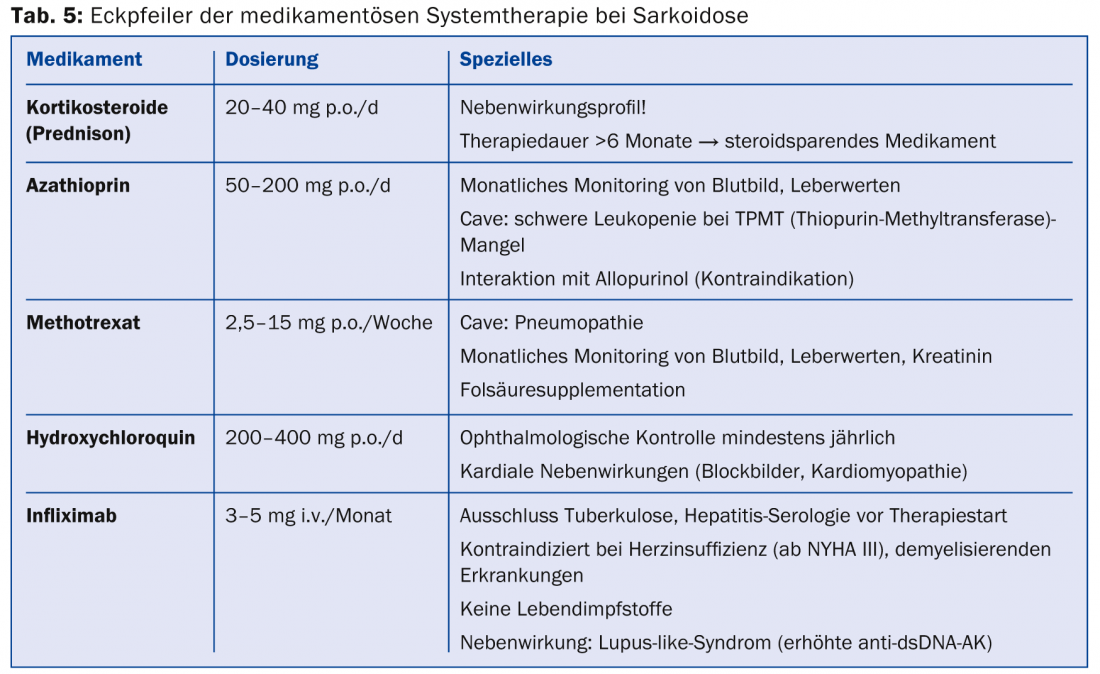
Debido a su buena y rápida eficacia, los corticosteroides son la primera elección. Se recomiendan dosis de 20-40 mg en dosificación gradual durante aproximadamente medio año, pero no existen ensayos controlados aleatorizados. Debe evaluarse precozmente el uso de sustancias ahorradoras de esteroides (azatioprina, metotrexato, leflunomida, micofenolato) para los efectos secundarios asociados a los esteroides. Sin embargo, el metotrexato debe prescribirse con precaución en la sarcoidosis pulmonar debido a la posibilidad de neumonitis por metotrexato. Dado que el TNF-α está implicado en la formación de granulomas, los antagonistas del TNF-α (especialmente el infliximab) son una buena terapia alternativa de segunda línea [34,35]. Se ha podido demostrar un beneficio sobre todo para la terapia de la sarcoidosis cutánea (lupus pernio), así como de las manifestaciones pulmonares y neurológicas. El fármaco antipalúdico hidroxicloroquina consigue buenos resultados en la sarcoidosis cutánea y en la hipercalcemia. En el síndrome de Löfgren, se establece una terapia sintomática con antiinflamatorios no esteroideos.
Literatura:
- Hunninghake GW, et al: Declaración de la ATS/ERS/WASOG sobre la sarcoidosis. Sociedad Torácica Americana/Sociedad Respiratoria Europea/Asociación Mundial de Sarcoidosis y otros Trastornos Granulomatosos. Sarcoidosis Vasc Diffuse Lung Dis 1999; 16(2): 149-173.
- Fernández FE: [Epidemiology of sarcoidosis]. Arch Bronconeumol 2007; 43(2): 92-100.
- Rybicki BA, Iannuzzi MC: Epidemiología de la sarcoidosis: avances recientes y perspectivas de futuro. Semin Respir Crit Care Med 2007; 28(1): 22-35.
- Pietinalho A, et al: La frecuencia de la sarcoidosis en Finlandia y Hokkaido, Japón. Un estudio epidemiológico comparativo. Sarcoidosis 1995; 12(1): 61-67.
- Rossman MD, Kreider ME: Lecciones aprendidas de ACCESS (A Case Controlled Etiologic Study of Sarcoidosis). Proc Am Thorac Soc 2007; 4(5): 453-456.
- Deubelbeiss U, et al.: La prevalencia de la sarcoidosis en Suiza está asociada a factores medioambientales. Eur Respir J 2010; 35(5): 1088-1097.
- Hosoda Y, et al.: Epidemiología mundial de la sarcoidosis. ¿Qué historia nos cuentan la prevalencia y la incidencia? Clin Chest Med 1997; 18(4): 681-694.
- Izbicki G, et al: Enfermedad pulmonar granulomatosa “similar al sarcoide” del World Trade Center en los trabajadores de rescate del Departamento de Bomberos de Nueva York. Tórax 2007; 131(5): 1414-1423.
- McGrath DS, et al: Epidemiología de la sarcoidosis familiar en el Reino Unido. Thorax 2000; 55(9): 751-754.
- Rossman MD, et al: HLA-DRB1*1101: un factor de riesgo significativo para la sarcoidosis en negros y blancos. Am J Hum Genet 2003; 73(4): 720-735.
- Berlin M, et al: El HLA-DR predice el pronóstico en pacientes escandinavos con sarcoidosis pulmonar. Am J Respir Crit Care Med 1997; 156(5): 1601-1605.
- Baughman RP, et al: Liberación del factor de necrosis tumoral por los macrófagos alveolares de pacientes con sarcoidosis. J Lab Clin Med 1990; 115(1): 36-42.
- Iida K, et al: Análisis de subconjuntos de células T y quimiocinas beta en pacientes con sarcoidosis pulmonar. Thorax 1997; 52(5): 431-437.
- Pinkston P, Bitterman PB, Crystal RG: Liberación espontánea de interleucina-2 por los linfocitos T pulmonares en la sarcoidosis pulmonar activa. N Engl J Med 1983; 308(14): 793-800.
- Judson MA: El diagnóstico de la sarcoidosis. Clin Chest Med 2008; 29(3): 415-427, viii.
- Baughman RP, et al: Características clínicas de los pacientes de un estudio de casos y controles de sarcoidosis. Am J Respir Crit Care Med 2001; 164(10 Pt 1): 1885-1889.
- Thomas KW, Hunninghake GW: Sarcoidosis. JAMA 2003; 289(24): 3300-3303.
- Siltzbach LE: Sarcoidosis: características clínicas y tratamiento. Med Clin North Am 1967; 51(2): 483-502.
- Scadding JG: Pronóstico de la sarcoidosis intratorácica en Inglaterra. Una revisión de 136 casos tras cinco años de observación. Br Med J 1961; 2(5261): 1165-1172.
- Chapman JT, Mehta AC: Broncoscopia en la sarcoidosis: intervenciones diagnósticas y terapéuticas. Curr Opin Pulm Med 2003; 9(5): 402-407.
- Shorr AF, Torrington KG, Hnatiuk OW. Biopsia endobronquial para la sarcoidosis: un estudio prospectivo. Tórax 2001; 120(1): 109-114.
- Koerner SK, et al: Biopsia pulmonar transbronquial para el diagnóstico de la sarcoidosis. N Engl J Med 1975; 293(6): 268-270.
- Gilman MJ, Wang KP: Biopsia pulmonar transbronquial en la sarcoidosis. Un enfoque para determinar el número óptimo de biopsias. Am Rev Respir Dis 1980; 122(5): 721-724.
- Mitchell DM, et al: Biopsia pulmonar transbronquial mediante broncoscopio de fibra óptica en el diagnóstico de la sarcoidosis. Br Med J 1980; 280(6215): 679-681.
- Agarwal R, et al: Eficacia y seguridad de la EBUS-TBNA con sonda convexa en la sarcoidosis: revisión sistemática y metaanálisis. Respir Med 2012; 106(6): 883-892.
- Costabel U: Relaciones CD4/CD8 en el líquido de lavado broncoalveolar: ¿valen para el diagnóstico de la sarcoidosis? Eur Respir J 1997; 10(12): 2699-2700.
- Zaiss AW, et al: [Relación T4/T8 en el líquido de lavado broncoalveolar: sensibilidad y especificidad para el diagnóstico de la sarcoidosis]. Prax Klin Pneumol 1988; 42 Suppl 1: 233-234.
- Chowdhury FU, et al: Reacción sarcoide a malignidad en la PET/TC integrada con (18)F-FDG de cuerpo entero: prevalencia y patrón de la enfermedad. Clin Radiol 2009; 64(7): 675-681.
- Ardeniz O, Cunningham-Rundles C: Enfermedad granulomatosa en la inmunodeficiencia variable común. Clin Immunol 2009; 133(2): 198-207.
- Khasnis AA, Calabrese LH: Inhibidores del factor de necrosis tumoral y enfermedad pulmonar: una paradoja de eficacia y riesgo. Semin Arthritis Rheum 2010; 40(2): 147-163.
- Goldberg HJ, et al: Sarcoidosis tras el tratamiento con interferón alfa: serie de casos y revisión de la literatura. Respir Med 2006; 100(11): 2063-2068.
- Studdy PR, Bird R: Enzima convertidora de angiotensina sérica en la sarcoidosis: su valor en la práctica clínica actual. Ann Clin Biochem 1989; 26(Pt 1): 13-18.
- Siltzbach LE, et al: Curso y pronóstico de la sarcoidosis en el mundo. Am J Med 1974; 57(6): 847-852.
- Judson MA, et al: Eficacia del infliximab en la sarcoidosis extrapulmonar: resultados de un ensayo aleatorizado. Eur Respir J 2008; 31(6): 1189-1196.
- Baughman RP, et al: Tratamiento con infliximab en pacientes con sarcoidosis crónica y afectación pulmonar. Am J Respir Crit Care Med 2006; 174(7): 795-802.
PRÁCTICA GP 2015; 10(3): 10-18









