La inmunofluorescencia directa e indirecta sigue siendo el patrón oro para la detección de anticuerpos y autoanticuerpos específicos en el penfigoide bulloso. Se recomiendan los esteroides tópicos de alta eficacia o los corticosteroides sistémicos como terapia de primera línea. En caso de falta de respuesta o contraindicaciones, pueden utilizarse fármacos inmunomoduladores e inmunosupresores. Según varios informes de casos, el omalizumab y el dupilumab han demostrado ser opciones de tratamiento eficaces y actualmente se están investigando otros productos biológicos.
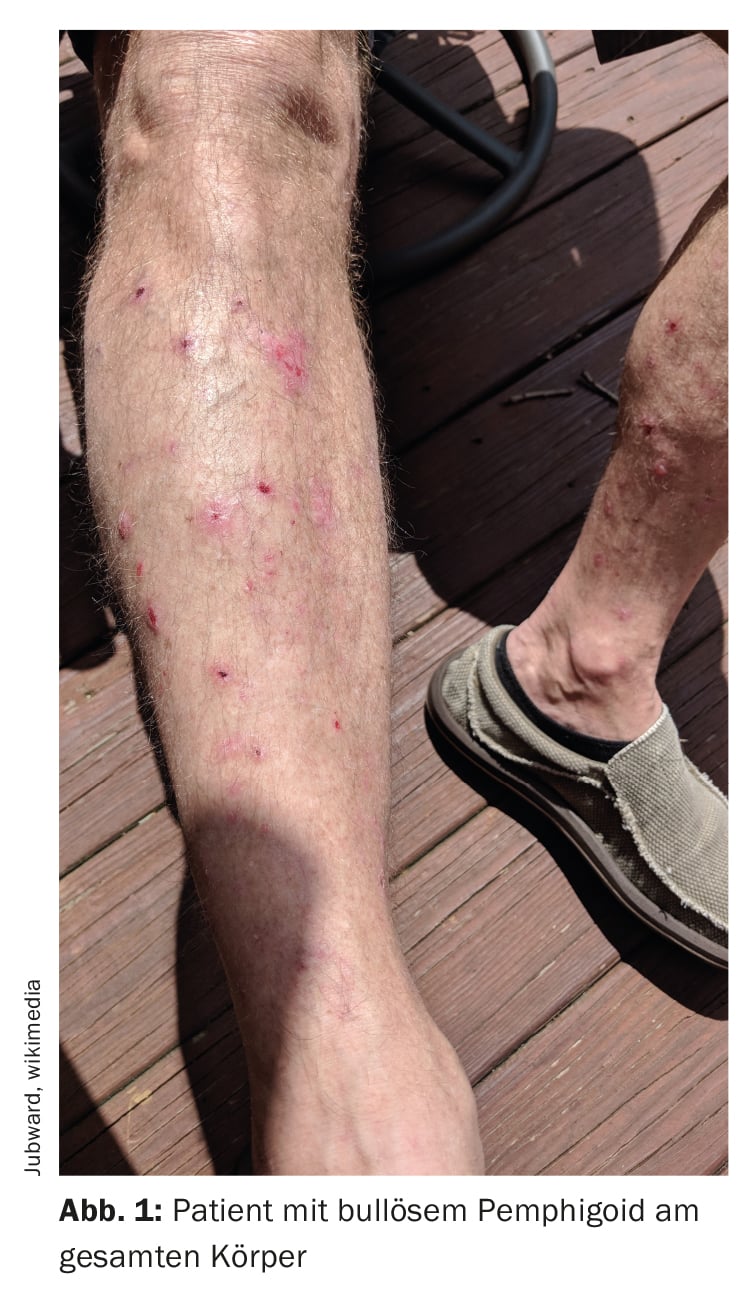
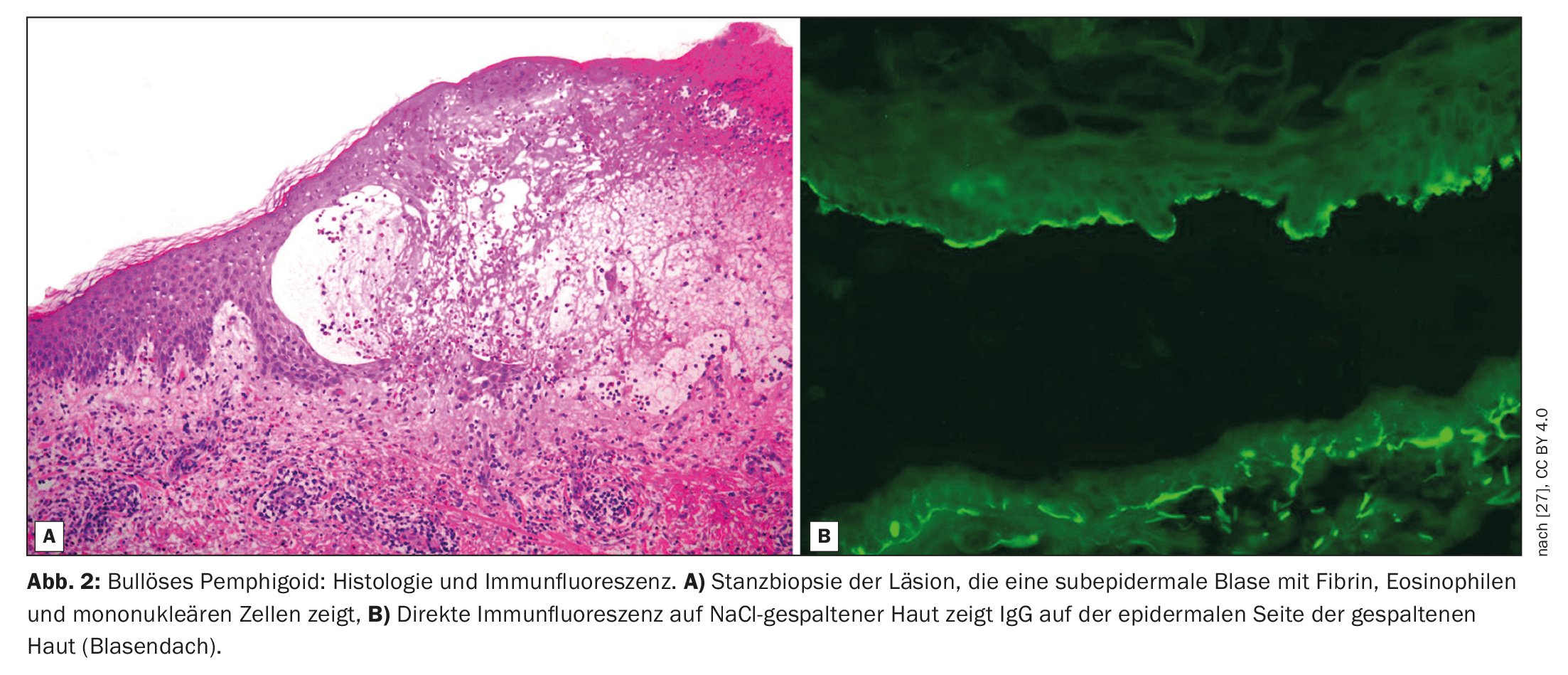
El penfigoide bulloso (PB) se caracteriza por la formación de ampollas subepidérmicas, es decir, ampollas dentro de la zona de unión dermoepidérmica (Fig. 1, Fig. 2) [1]. Se trata de la dermatosis bullosa autoinmune más frecuente en Suiza y Europa, según informó el Prof. Dr. Luca Borradori, director clínico y médico jefe de la Clínica Universitaria de Dermatología del Inselspital de Berna [2]. En las últimas décadas, también se ha observado un aumento de la incidencia de la PA en este país. Entre las posibles causas se encuentran el envejecimiento de la población, la asociación con enfermedades neurológicas cada vez más comunes y ciertos medicamentos, así como un mayor conocimiento de las variantes atípicas sin ampollas [28]. El tratamiento de la PA tiene como objetivo controlar la enfermedad, aliviar los síntomas de picor y mejorar la calidad de vida de los afectados. Los métodos de tratamiento dependen principalmente de la gravedad de la enfermedad. Debe prestarse atención a un perfil favorable de riesgo-beneficio del tratamiento respectivo, afirma el Prof. Borradori.
Sospecha clínica de PA: la serología aporta certeza
El diagnóstico de la PA se basa en una combinación de criterios, que incluyen las características clínicas, los hallazgos en el microscopio óptico y la detección de autoanticuerpos en la piel mediante inmunofluorescencia directa (IFD) [3]. La manifestación clínica clásica de la PA es una erupción muy pruriginosa con ampollas generalizadas (ampollas abultadas y pruriginosas de diversos tamaños llenas de líquido seroso). Sin embargo, en los estadios iniciales o en las variantes atípicas, pueden presentarse únicamente lesiones excoriadas, eccematosas o urticariales localizadas o generalizadas [4]. Las mucosas se ven afectadas en un 10-30%. El hallazgo típico en la inmunofluorescencia directa es la detección de inmunoglobulina (Ig)G lineal unida a la zona de unión dermoepidérmica, más raramente también IgM e IgA. Estos autoanticuerpos se unen principalmente a las proteínas BP180 y BP230, que son componentes de los hemidesmosomas. Representan una importante conexión celular y tienen una función esencial para la estabilidad mecánica de la piel [1].
Para confirmar la sospecha diagnóstica de PA, en las directrices s2K de un grupo de expertos de la EADV, actualizadas en 2022, se recomiendan los siguientes pasos adicionales de aclaración diagnóstica de laboratorio [3]:
- Detección de autoanticuerpos IgG circulantes contra la zona de la membrana basal (ZMB) mediante microscopía de inmunofluorescencia indirecta (IIF) utilizando piel humana normal escindida con solución de NaCl.
- Detección de autoanticuerpos IgG anti-BP180-NC16A y/o autoanticuerpos IgG anti-BP230 mediante ELISA (ensayo inmunoenzimático).
- Las novedosas pruebas “multivariantes” también se recomiendan para la detección de autoanticuerpos IgG anti-BMZ circulantes. Estos ensayos de mosaico BIOCHIP combinan diferentes sustratos antigénicos. En los raros casos de PA en los que los anticuerpos circulantes anti-BMZ no son detectables ni por microscopía de inmunofluorescencia indirecta ni por ELISA comercial, se recomienda realizar pruebas adicionales para aumentar la sensibilidad diagnóstica y descartar otras enfermedades autoinmunes de la unión dermoepidérmica (en particular el penfigoide anti-P200 o la epidermólisis bullosa adquirida). Encontrará un resumen de las pruebas correspondientes en las directrices s2K publicadas en JEADV [3].
Fisiopatología: también intervienen mecanismos independientes del complemento
Las relaciones fisiopatológicas en la PA son muy complejas [4]. Por un lado, la activación del sistema del complemento con la producción de anafilatoxinas y la activación de la respuesta inmunitaria innata con el consiguiente reclutamiento y activación de basófilos, eosinófilos, neutrófilos, monocitos/macrófagos y mastocitos desempeñan un papel clave en la PA [5,6, 28-31]. Por otro lado, en los últimos años los investigadores han descubierto cada vez más mecanismos inflamatorios independientes del complemento en relación con los anticuerpos de la PA. Los queratinocitos, en particular, parecen ser capaces de secretar un gran número de citocinas proinflamatorias patogénicamente relevantes [4,7,32].
Un estudio demostró que tanto la BP-IgG como la BP-IgE son capaces de unirse directamente a la superficie de queratinocitos humanos cultivados, con la consiguiente pérdida de hemidesmosomas en la zona de la membrana basal [8]. Los eosinófilos están implicados en la patogénesis de la PA al mediar en los efectos de los anticuerpos IgE anti-BP180 y contribuir a la separación dermoepidérmica. En presencia de IgG o IgE, los eosinófilos pueden mediar directamente en la separación dermoepidérmica [9,10]. Freire et al. encontraron niveles elevados de IgE tanto anti-BP180 como anti-BP230 mediante ELISA e inmunofluorescencia en comparación con los controles sanos [11].
Los autoanticuerpos IgE anti-BP180 están presentes en la mayoría de los pacientes con PA, y sus niveles se correlacionan con la actividad de la enfermedad [12–15]. Además, los estudios de inmunofluorescencia directa han demostrado que la mayoría de los pacientes con PA tenían células IgE+ en la piel, en contraste con los controles sanos. En general, las pruebas sugieren que existe una vía adicional independiente del complemento, dependiente de Th2 y mediada por eosinófilos que contribuye al daño tisular y a las características clínicas en la PA [4].
| “Tratar hasta el objetivo” con supervisión y seguimiento de los progresos |
| – El grupo de expertos de la EADV recomienda en general una duración del tratamiento de entre 9 y 12 meses para la PA [3]. |
| – Se recomienda interrumpir el tratamiento en pacientes que hayan estado libres de síntomas durante al menos 1-6 meses con una terapia mínima con prednisona oral (0,1 mg/kg/día), propionato de clobetasol (10 g/semana) o inmunosupresores. La interrupción del tratamiento también se ve influida por el estado general del paciente y la presencia de ciertas comorbilidades. |
| – ELISA anti-BP180 (es decir, >27 U/mL según lo determinado por la prueba MBL**) y, en menor medida, los estudios DIF$ han informado de que tienen valor predictivo para la aparición de recaídas tras la interrupción del tratamiento [24]. Puede que merezca la pena considerar el uso de estas pruebas antes de interrumpir el tratamiento. |
| – Además, debe prestarse atención a la posible insuficiencia suprarrenal causada por el uso de corticosteroides exógenos (CS) (también tras el uso de CS tópicos). |
| Se recomienda un examen de seguimiento en el tercer mes tras la interrupción del tratamiento. Este periodo parece ser suficiente para reconocer la mayoría de las recaídas de la PA [24–26]. Independientemente de ello, la reaparición de picores, excoriaciones y/o lesiones cutáneas inflamatorias debe ser siempre aclarada por un médico. |
| ** MBL= Laboratorios Médicos y Biológicos $ DIF=Inmunofluorescencia directa |
Nuevas opciones de tratamiento: Los biológicos como terapia alternativa
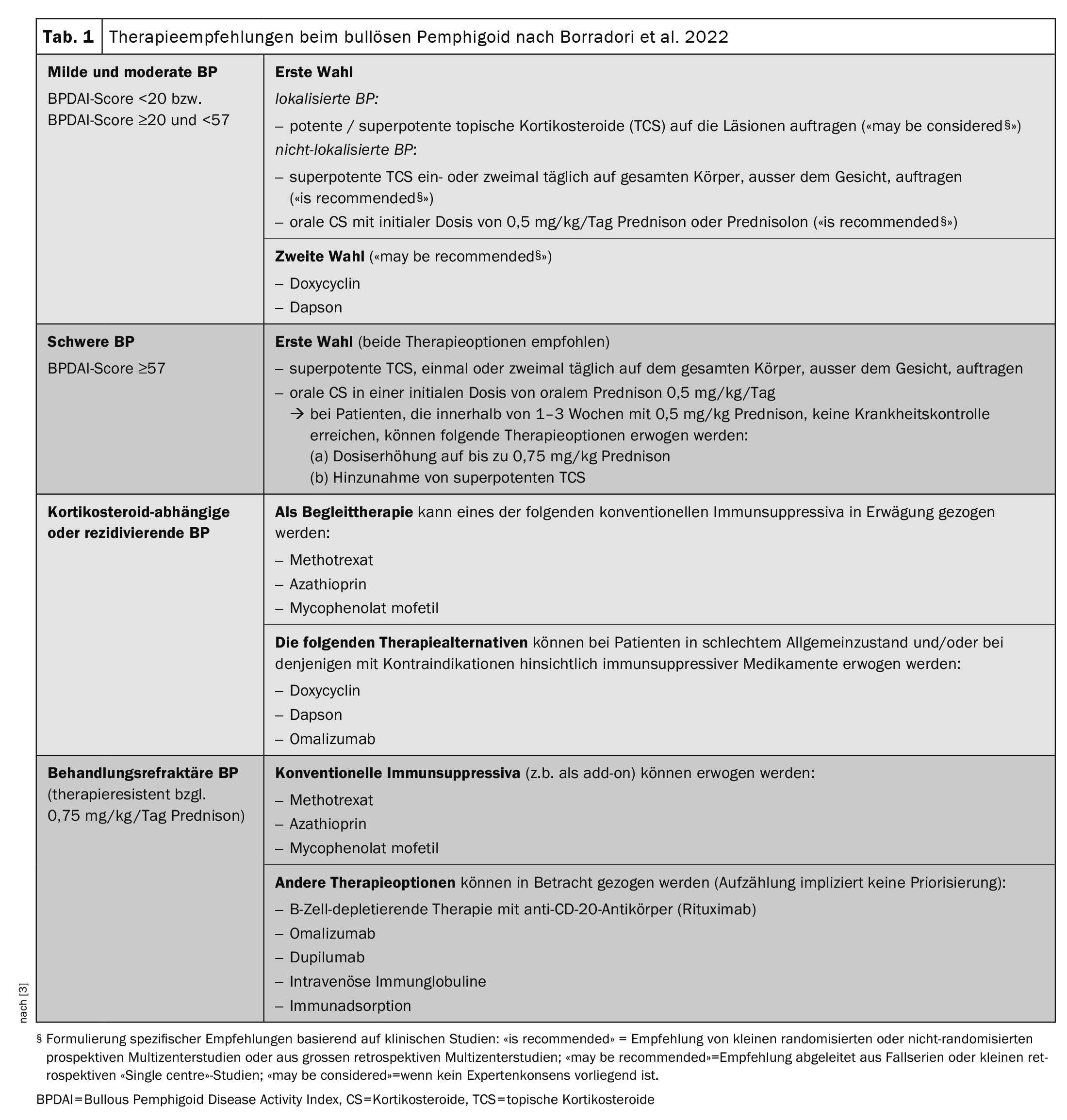
La tabla 1 [3] ofrece una visión general de las opciones de tratamiento disponibles en la actualidad. Las directrices siguen recomendando el uso de corticosteroides tópicos superpotentes (TCS) o esteroides sistémicos (prednisolona 0,5 mg/kg/d) como tratamiento de primera línea. En muchos casos, los síntomas pueden contenerse de este modo, pero “hay que tener paciencia”, explicó el profesor Borradori [2]. La duración estimada del tratamiento es de 9 a 12 meses. Sin embargo, también hay pacientes para los que la terapia estándar no es eficaz o está contraindicada. “Necesitamos terapias buenas, nuevas y eficaces”, subrayó el ponente [2]. La observación de que los pacientes con PA suelen presentar niveles séricos elevados de IgE y autoanticuerpos circulantes IgE específicos de BP180- y BP230 apoya la idea de que la IgE desempeña un papel en la patogénesis de la PA [16,17]. El uso de omalizumab, un anticuerpo monoclonal humanizado que inhibe la unión de la IgE a su receptor de alta afinidad (FcεRI), es por tanto una elección obvia. Los informes de casos muestran que el tratamiento con omalizumab provoca una fuerte disminución de la expresión de FcεRI en los basófilos circulantes y una fuerte reducción de las células FcεRI + en la piel de los pacientes tratados [16].
Otras terapias biológicas dirigidas que se han mostrado prometedoras en la PA incluyen el dupilumab, un anticuerpo monoclonal IgG4 humano que se une a la subunidad α del receptor de IL-4, inhibiendo así las vías de señalización IL-4/IL-13 [18–21]. “Tenemos muchas series de casos que demuestran que el dupilumab tiene un buen efecto en pacientes con penfigoide bulloso”, informó el profesor Borradori [19,22]. El biológico demostró ser eficaz tanto como monoterapia como en combinación con una terapia estándar. Tras el éxito de las pruebas clínicas de fase II, actualmente se está investigando la eficacia y la seguridad del dupilumab en la PA en el ensayo de fase III LIBERTY-BP (NCT04206553) [23].
Congreso: Formación avanzada conjunta BE-BS-ZH
Literatura:
- Diagnóstico y tratamiento del pénfigo vulgar/foliáceo y del penfigoide bulloso, directriz AWMF S2k 2019, Sociedad Dermatológica Alemana e.V. (DDG), registro AWMF nº: 013-071.
- “Terapia del penfigoide bulloso: procedimiento práctico”, Prof. Dr. Luca Borradori, Formación continua conjunta de las clínicas dermatológicas de Berna, Basilea, Zúrich, Inselspital de Berna, 25.05.2023.
- Borradori L, et al: Directrices S2 K actualizadas para el tratamiento del penfigoide ampolloso iniciadas por la Academia Europea de Dermatología y Venereología (EADV). J Eur Acad Dermatol Venereol 2022; 36(10): 1689-1704.
- Cole C, et al: Insights into the pathogenesis of bullous pemphigoid: The role of complement-independent mechanisms. Compass Dermatol 2022; 10 (4): 171-180.
- Liu Z, et al: La formación de ampollas subepidérmicas inducida por autoanticuerpos humanos frente a BP180 requiere agentes inmunitarios innatos en un modelo de ratón humanizado de penfigoide bulloso. J Autoimmun 2008; 31(4): 331-338.
- Liu Z, et al: Sinergia entre una cascada de plasminógeno y la MMP-9 en la enfermedad autoinmune. J Clin Invest 2005; 115(4): 879-887.
- Bao L, et al: La reactividad subunidad-específica de los autoanticuerpos contra la laminina-332 revela mecanismos inflamatorios directos en los queratinocitos. Front Immunol 2021; 12: 775412.
- Messingham KN, et al: Efectos independientes del FcR de los autoanticuerpos IgE e IgG en el penfigoide bulloso. J I 2011; 187(1): 553-560.
- Amber KT, et al: El papel de los eosinófilos en el penfigoide bulloso: un modelo en desarrollo de la patogenicidad de los eosinófilos en la enfermedad mucocutánea. Front Med (Lausana). 2018; 5: 201.
- de Graauw E, et al: Evidencia de un papel de los eosinófilos en la formación de ampollas en el penfigoide bulloso. Alergia 2017; 72(7): 1105-1113.
- Freire PC, Munoz CH, Stingl G: Autoreactividad IgE en el penfigoide bulloso: los eosinófilos y los mastocitos como dianas principales de los reactivos inmunitarios patógenos. Br J Dermatol 2017; 177(6): 1644-1653.
- Hashimoto T, et al: Detección de autoanticuerpos IgE frente a BP180 y BP230 y su relación con las características clínicas en el penfigoide bulloso. Br J Dermatol 2017; 177(1): 141-151.
- van Beek N, et al: Correlación de los niveles séricos de autoanticuerpos IgE contra BP180 con la actividad de la enfermedad del penfigoide ampolloso. JAMA Dermatol 2017; 153(1): 30-38.
- Bing L, et al: Levels of Anti-BP180 NC16A IgE do not Correlate With Severity of Disease in the Early Stages of Bullous Pemphigoid. Arch Dermatol Res 2015; 307(9): 849-854.
- Salz M, et al: Los niveles séricos elevados de IL-31 en pacientes con penfigoide bulloso se correlacionan con el número de eosinófilos y se asocian con la BP180-IgE. J Dermatol Sci 2017; 87(3): 309-311.
- Seyed Jafari SM, et al: Efectos del omalizumab sobre la expresión de FcεRI e IgE en la piel lesional del penfigoide bulloso. Front Immunol 2019; 10: 1919.
- Messingham KN, Crowe TP, Fairley JA: La intersección de los autoanticuerpos IgE y la eosinofilia en la patogénesis del penfigoide bulloso. Front Immunol. 2019;10: 2331.
- Abdat R, et al: Dupilumab como terapia novedosa para el penfigoide bulloso: una serie de casos multicéntricos. J Am Acad Dermatol 2020; 83(1): 46-52.
- Zhang Y, et al: Eficacia y seguridad del dupilumab en el penfigoide bulloso de moderado a grave. Front Immunol 2021; 12: 738907.
- Seyed Jafari SM, et al: Informe de un caso: Combinación de omalizumab y dupilumab para el penfigoide bulloso recalcitrante. Front Immunol 2020; 11: 611549.
- Kaye A, et al: Dupilumab para el tratamiento del penfigoide bulloso recalcitrante. JAMA Dermatol 2018; 154(10): 1225-1256.
- Klepper EM, Robinson HN: Dupilumab para el tratamiento del penfigoide ampolloso inducido por nivolumab: informe de un caso y revisión de la literatura. Dermatol Online J. 2021;27(9): 1-6.
- Biblioteca Nacional de Medicina de EE.UU. Un estudio para evaluar la eficacia y seguridad del dupilumab en pacientes adultos con penfigoide bulloso (LIBERTY-BP). Bethesda, MD: Biblioteca Nacional de Medicina de EE.UU. (2019).
- Bernard P, et al: Factores de riesgo de recaída en pacientes con penfigoide bulloso en remisión clínica: un estudio multicéntrico, prospectivo y de cohortes. Arch Dermatol 2009; 145: 537-542.
- Joly P, et al: Comparación de dos regímenes de corticosteroides tópicos en el tratamiento de pacientes con penfigoide bulloso: un estudio multicéntrico aleatorizado. J Invest Dermatol 2009; 129: 1681-1687.
- Fichel F, et al: Factores clínicos e inmunológicos asociados a la recaída del penfigoide bulloso durante el primer año de tratamiento: un estudio multicéntrico y prospectivo. JAMA Dermatol 2014; 150: 25-33.
- Michelerio A, Tomasini C: Ampollas y milia alrededor del catéter de diálisis peritoneal: Un caso de penfigoide bulloso localizado. Dermatopatología 2022; 9(3): 282-286. www.mdpi.com/2296-3529/9/3/33,(última consulta: 10/08/2023).
- Holtsche MM, Boch K, Schmidt E: J Dtsch Dermatol Ges 2023; 21(4): 405-413.
- Chen R, et al: J Clin Invest 2001; 108(8): 1151-1158.
- Nelson KC, et al: J Clin Invest 2006; 116(11): 2892-900.
- Leighty L, et al: Arch Dermatol Res 2007; 299(9): 417-422.
- Schmidt E, et al: J Invest Dermatol 2000; 115(5): 842-848.
DERMATOLOGIE PRAXIS 2023; 33(5): 50-52 (publicado el 30.10.23, antes de impresión)