Las dermatosis bullosas autoinmunes representan un grupo heterogéneo de enfermedades autoinmunes raras, a veces graves, que incluyen el pénfigo y las enfermedades penfigoides, la epidermólisis bullosa adquirida y la dermatitis herpetiforme Duhring. Una característica común de las dermatosis autoinmunes bullosas -con la excepción de la enfermedad de Duhring- son los autoanticuerpos dirigidos contra proteínas estructurales de la piel y las mucosas y responsables de una pérdida de la integridad cutánea [1].
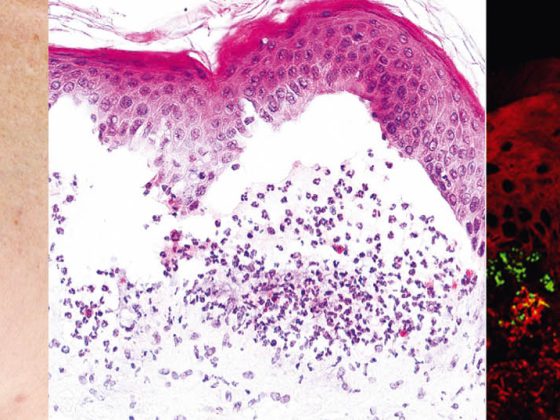
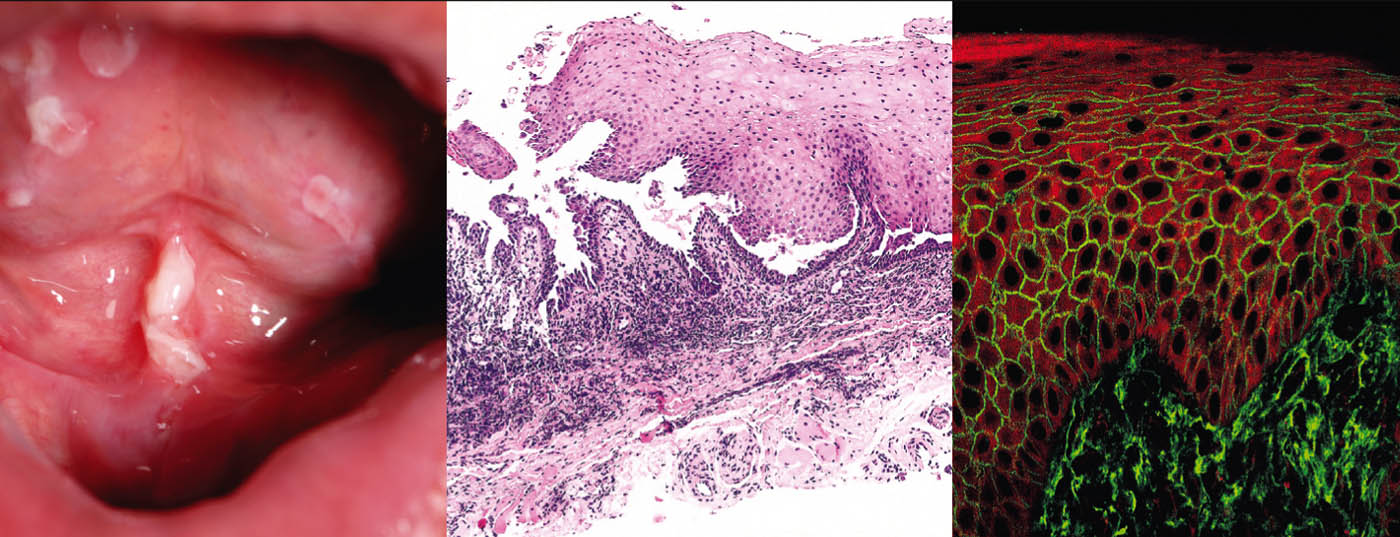
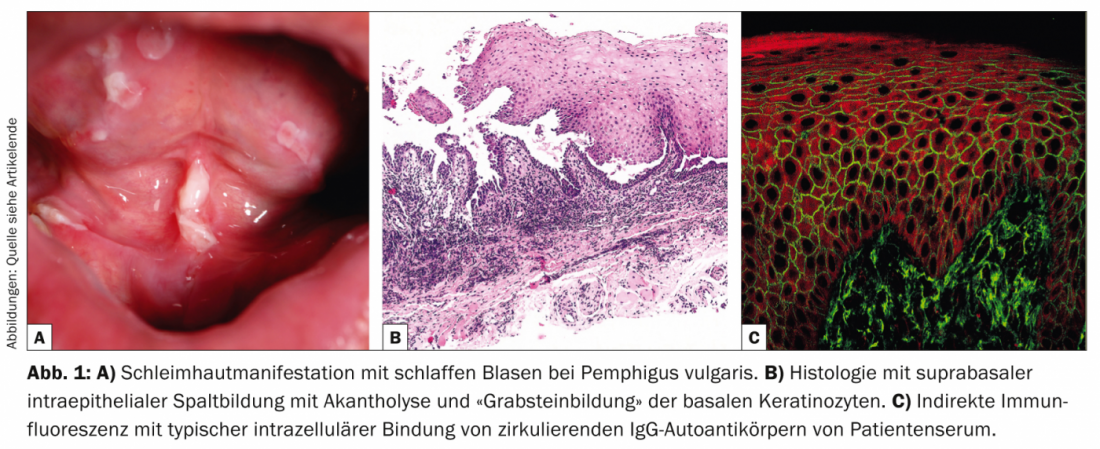
El pénfigo vulgar (Tab. 1, Fig. 1) se manifiesta con ampollas generalizadas, mucocutáneas y flácidas que se rompen muy rápidamente, por lo que el cuadro clínico suele estar dominado por erosiones y costras.

La afectación de las mucosas, especialmente de la boca, que es muy dolorosa, se produce en la mayoría de los pacientes, por lo que no es infrecuente que el diagnóstico lo realicen inicialmente los dentistas. Desde el punto de vista patogénico, el pénfigo vulgar se caracteriza por la presencia de autoanticuerpos (AK) contra las proteínas de adhesión celular: las desmogleínas 1 y 3. En los pacientes con infestación exclusivamente oral, los AK se dirigen contra la desmogleína 3. Histopatológicamente, hay formación de hendiduras intraepidérmicas suprabasales con acantólisis y “formación de lápida” de los queratinocitos basales. La inmunofluorescencia directa (IFD) muestra depósitos intercelulares típicos de IgG y C3 en la epidermis. La inmunofluorescencia indirecta (IIF) del suero del paciente confirma la circulación de autoanticuerpos IgG. Mediante ELISA, se puede lograr la detección directa de desmogleína 3 y desmogleína 1 en el suero de los pacientes [2].
El objetivo principal de la terapia es reducir la producción de autoanticuerpos. Los corticosteroides sistémicos y otros inmunosupresores como la azatioprina, el mofetil micofenolato, la ciclofosfamida o la ciclosporina siguen siendo las terapias estándar. Otras opciones son la plasmaféresis o las inmunoglobulinas intravenosas (IGIV). En los casos resistentes a la terapia, el anticuerpo anti-CD20 rituximab en particular es una alternativa prometedora. Además del pénfigo vulgar, el grupo de enfermedades del pénfigo también incluye el pénfigo foliáceo [3], el pénfigo herpetiforme [4], el pénfigo paraneoplásico [5] y el pénfigo IgA.
Penfigoide bulloso
Con una incidencia de 12,1 nuevos casos/millón/año en Suiza, el penfigoide ampolloso (Tab. 2, Fig. 2) es la enfermedad más frecuente del grupo del penfigoide y al mismo tiempo la dermatosis autoinmune formadora de ampollas más frecuente de todas [6].

El penfigoide bulloso suele aparecer en la vejez y se caracteriza por ampollas abultadas en la piel inflamada-enrojecida o normal que pican intensamente. A menudo, esta enfermedad cursa inicialmente sin ampollas y se diagnostica como eccema, urticaria o prurigo debido al pronunciado prurito. Desde el punto de vista patogénico, la enfermedad está causada por autoanticuerpos contra el PB 180 (también conocido como colágeno de tipo XVII). La histología muestra ampollas subepidérmicas con epidermis intacta e infiltrados prominentes ricos en eosinófilos. En la IFD de la piel perilesional, los depósitos de IgG se encuentran a lo largo de la zona de unión dermoepidérmica (membrana basal), lo que puede confirmarse en la IFI con la detección de anticuerpos IgG que se unen a la membrana basal. El nivel sérico de autoanticuerpos contra el BP180 se correlaciona con la actividad de la enfermedad y puede determinarse durante el curso para determinar la necesidad de terapia adicional.
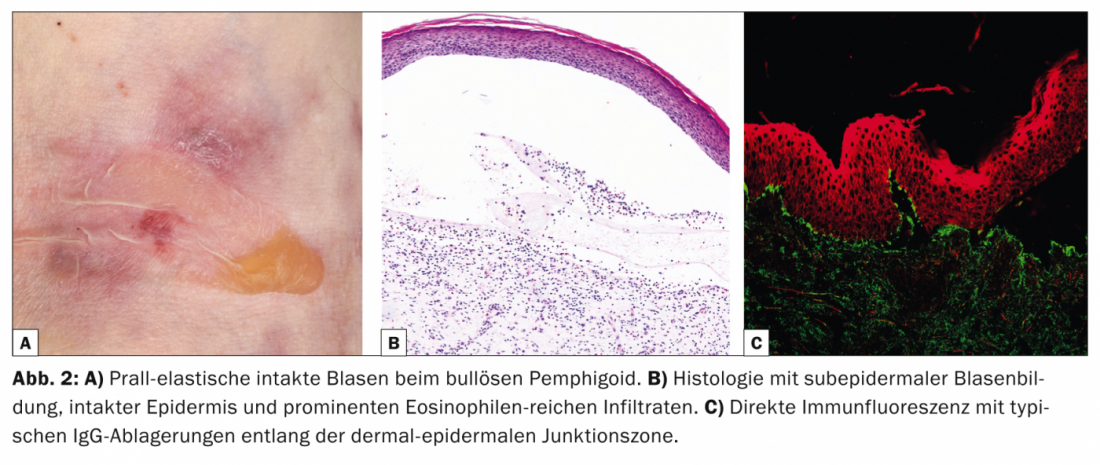
Los corticosteroides tópicos o sistémicos siguen siendo la base de la terapia. Otras opciones son las tetraciclinas con nicotinamidas, la dapsona o, en la enfermedad grave, los inmunosupresores, especialmente la azatioprina, así como la IGIV y el rituximab en los casos resistentes a la terapia.
IgA lineal, dermatosis bullosa
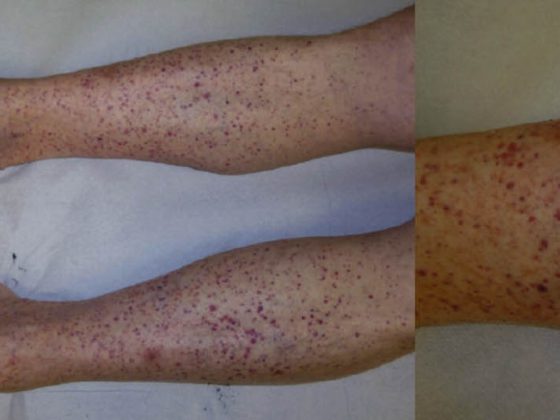
La dermatosis IgA lineal (LABD, Tab. 3, Fig. 3) se caracteriza por vesículas y bullas pruriginosas, generalizadas y acentuadas en el lado extensor. La enfermedad se da tanto en adultos como en niños.

En la infancia, es la dermatosis bullosa autoinmune más común y suele ser autolimitada [7]; en los adultos, la LABD suele estar asociada a fármacos (vancomicina) [8]. Histológicamente, la LABD es similar a la enfermedad de Duhring en que se caracteriza por ampollas subepidérmicas, epidermis intacta e infiltrados ricos en neutrófilos en la unión dermoepidérmica. La DIF muestra depósitos lineales de IgA en la zona de la membrana basal.
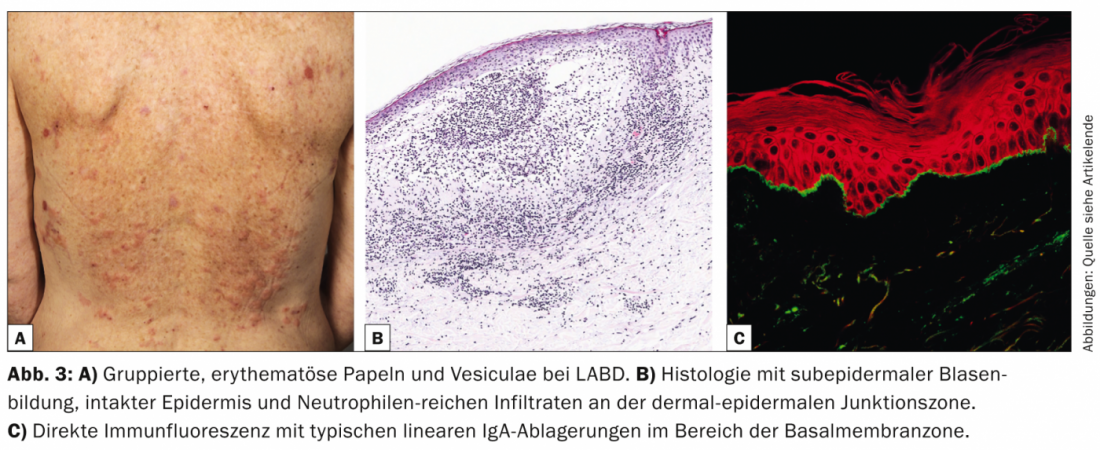
La IIF puede confirmar el diagnóstico detectando autoanticuerpos IgA circulantes que se unen al techo de la vejiga. La mayoría de los pacientes responden a la terapia con dapsona o sulfapiridinas. También se han descrito tratamientos exitosos en niños y adultos con diversos antibióticos (cicloxacilina, eritromicina, tetraciclina o trimetoprim-sulfametoxazol).
Epidermiolisis bullosa aquisita
La epidermiolisis bullosa aquística (EBA, Tab. 4, Fig. 4) se divide clínicamente en una forma mecano-bullosa localizada, a menudo acralada, no inflamatoria y cicatricial, y una variante generalizada, inflamatoria y no cicatricial. La enfermedad aparece sobre todo en la edad adulta media y es muy rara en niños.

Existe una estrecha asociación con la enfermedad inflamatoria intestinal [9]. Patogénicamente, la EBA se caracteriza por el depósito de autoanticuerpos IgG contra el procolágeno de tipo VII de la fibrilla de anclaje. Histológicamente, la muestra rutinaria muestra una ampolla subepidérmica con la epidermis intacta. En la DIF de la piel perilesional, los anticuerpos IgG se encuentran en bandas a lo largo de la zona de unión dermoepidérmica. En la IIF en piel humana separada por NaCl, que es positiva en el 50% de los casos, los autoanticuerpos IgG circulantes o, con menor frecuencia, los IgA se unen en el suelo de la vejiga.
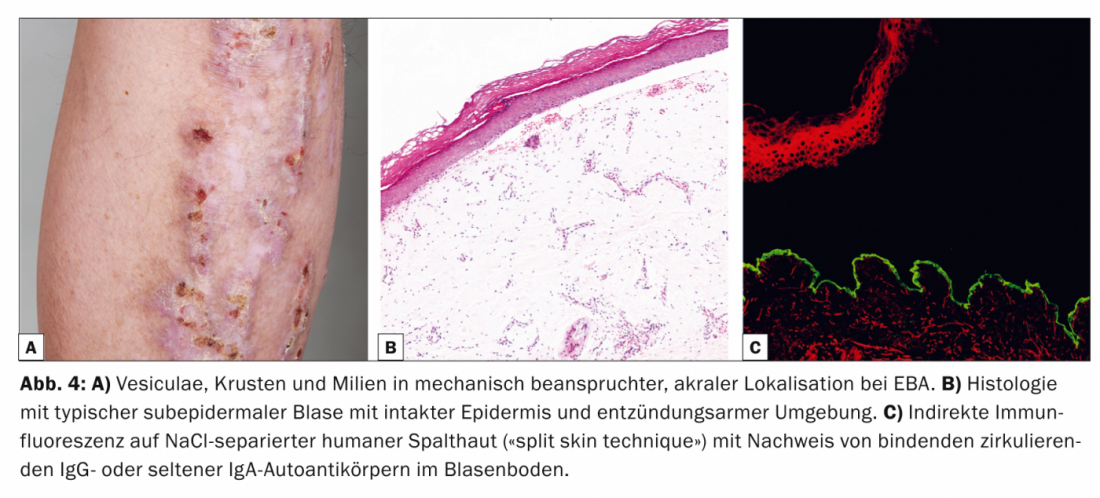
El Western blot y el ELISA pueden utilizarse de forma complementaria para detectar autoanticuerpos IgG circulantes [10]. La terapia de los TME es difícil, a menudo insatisfactoria y persigue principalmente un efecto inmunosupresor. Por lo tanto, las opciones de tratamiento actuales son principalmente los corticosteroides sistémicos, la dapsona o la colchicina. Además, existen informes de una respuesta positiva al anticuerpo anti-CD20 rituximab en casos resistentes al tratamiento.
Dermatitis herpetiforme
Dermatitis herpetiforme (DH, Tab. 5, Fig. 5) es una manifestación cutánea poco frecuente de la enteropatía sensible al gluten (enfermedad celíaca) y se presenta con pápulas eritematosas y papulovesículas con intenso picor, que aparecen principalmente en el lado extensor y en el sacro.

Desde el punto de vista patogénico, tanto la DH como la enfermedad celíaca están asociadas al genotipo HLA-DQ2, en el que están presentes autoanticuerpos IgA contra la reticulina, el endomisio y la transglutaminasa tisular, respectivamente. transglutaminasa epidérmica. Histológicamente, hay formación de hendiduras subepidérmicas e infiltrados ricos en neutrófilos con formación parcial de microabscesos [11].
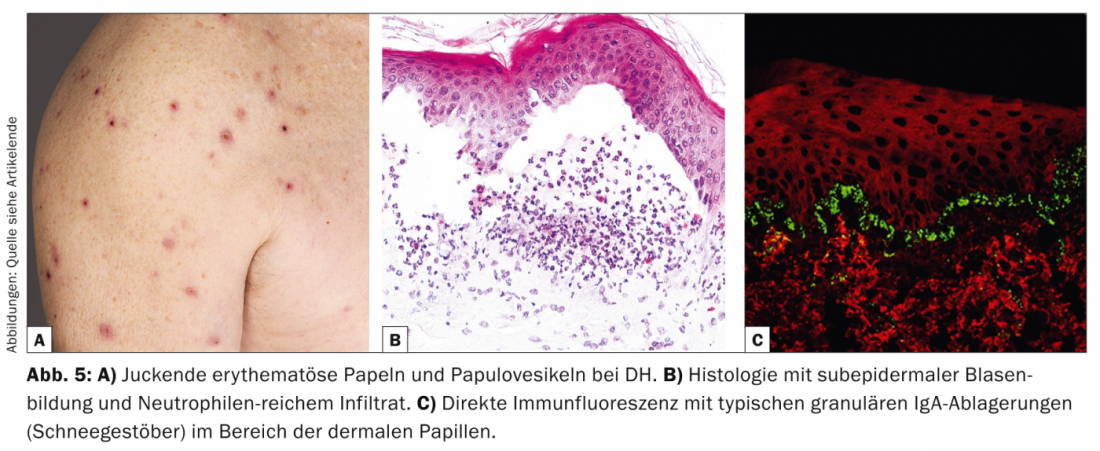
Histológicamente, la DH parece similar a la dermatosis bullosa IgA lineal, pero en la DIF de la piel perilesional se encuentran depósitos granulares de IgA (copos de nieve) en la zona de las papilas dérmicas. Desde el punto de vista terapéutico, ocupa un lugar destacado la dieta sin gluten, que mejora tanto las molestias gastroenterológicas como los síntomas cutáneos [12]. Las opciones alternativas son la dapsona, las sulfapiridinas o los corticosteroides sistémicos. La dapsona mejora las lesiones cutáneas pero no los síntomas intestinales.
Dra. Emmanuella Guenova, MD
Fuentes de las imágenes:
Imágenes clínicas: Archivo fotográfico del Hospital Universitario de Zúrich
Imágenes histológicas: Dra. Emmanuella Guenova, MD
Ilustraciones de inmunofluorescencia: Birgit Fehrenbacher
Literatura:
- Bolognia, JL, Jorizzo JL, Schaffer JV: Dermatología. 2012: Elsevier Health Sciences UK.
- Chan LS: Enfermedades ampollosas de la piel. 2009: Taylor & Francis.
- Guenova E, et al: Tinea incognito oculta bajo un pénfigo foliáceo aparentemente resistente al tratamiento. Acta Derm Venereol 2008; 88(3): 276-277.
- Lebeau S, et al: Pénfigo herpetiforme: análisis del perfil de autoanticuerpos durante el curso de la enfermedad con cambios en el fenotipo clínico. Clin Exp Dermatol 2010; 35(4): 366-372.
- Heizmann M, et al: Tratamiento satisfactorio del pénfigo paraneoplásico en el LNH folicular con rituximab: informe de un caso y revisión del tratamiento del pénfigo paraneoplásico en el LNH y la LLC. Am J Hematol 2001; 66(2): 142-144.
- Marazza G, et al: Incidencia del penfigoide bulloso y el pénfigo en Suiza: un estudio prospectivo de 2 años. Br J Dermatol 2009; 161(4): 861-868.
- de las Heras MN: Dermatosis bullosa IgA lineal de la infancia: buena respuesta al tratamiento antibiótico. Clin Exp Dermatol 2014; 39(3): 395-397.
- Tashima S, et al: Un caso de dermatosis bullosa IgA lineal inducida por vancomicina con anticuerpos IgA circulantes contra el dominio NC16a del BP180. Int J Dermatol 2014; 53(3): 207-209.
- Hundorfean G, Neurath MF, Sitaru C: Autoinmunidad contra el colágeno tipo VII en la enfermedad inflamatoria intestinal. J Cell Mol Med 2010; 14(10): 2393-2403.
- Calabresi V, et al: Sensibilidad de diferentes ensayos para el diagnóstico serológico de la epidermólisis ampollosa adquirida: análisis de una cohorte de 24 pacientes italianos. J Eur Acad Dermatol Venereol 2014; 28(4): 483-490.
- Hall MA, Lanchbury JS, Ciclitira PJ: Genes de la región HLA de clase II y susceptibilidad a la dermatitis herpetiforme: las asociaciones de DPB1 y TAP2 son secundarias a las de la subregión DQ. Eur J Immunogenet 1996; 23(4): 285-296.
- Hervonen K, et al: Dermatitis herpetiforme en niños: un estudio de seguimiento a largo plazo. Br J Dermatol 2014 [Epub ahead of print].
CONCLUSIÓN PARA LA PRÁCTICA
- En todas las dermatosis bullosas autoinmunes, además de la historia clínica, es importante el examen de todo el tegumento, incluida la piel. La inspección de las mucosas y las uñas es obligatoria.
- Si se sospecha clínica o histológicamente una enfermedad bullosa autoinmune, el diagnóstico debe confirmarse mediante la detección de los autoanticuerpos subyacentes (tinción de anticuerpos unidos a tejido en inmunofluorescencia directa en sección de tejido o detección en suero mediante inmunofluorescencia indirecta o ELISA).
- En la terapia de las dermatosis bullosas autoinmunes se siguen utilizando los inmunosupresores clásicos (por ejemplo, corticosteroides, azatioprina).
- Las opciones terapéuticas más recientes son los anticuerpos anti-CD20 (Rituximab), que conducen a una reducción de los autoanticuerpos.
PRÁCTICA DERMATOLÓGICA 2014; 24(4): 6-10