Los estudios han demostrado que la alteración del transporte de células inmunitarias y las células inmunitarias patógenas son factores cruciales responsables de la inflamación de la mucosa y la destrucción del tejido en la EII. Una barrera intestinal defectuosa y la disbiosis microbiana conducen a dicha acumulación y a la activación local de las células inmunitarias, lo que da lugar a un bucle de citoquinas proinflamatorias que anula las señales antiinflamatorias y provoca una inflamación intestinal crónica.
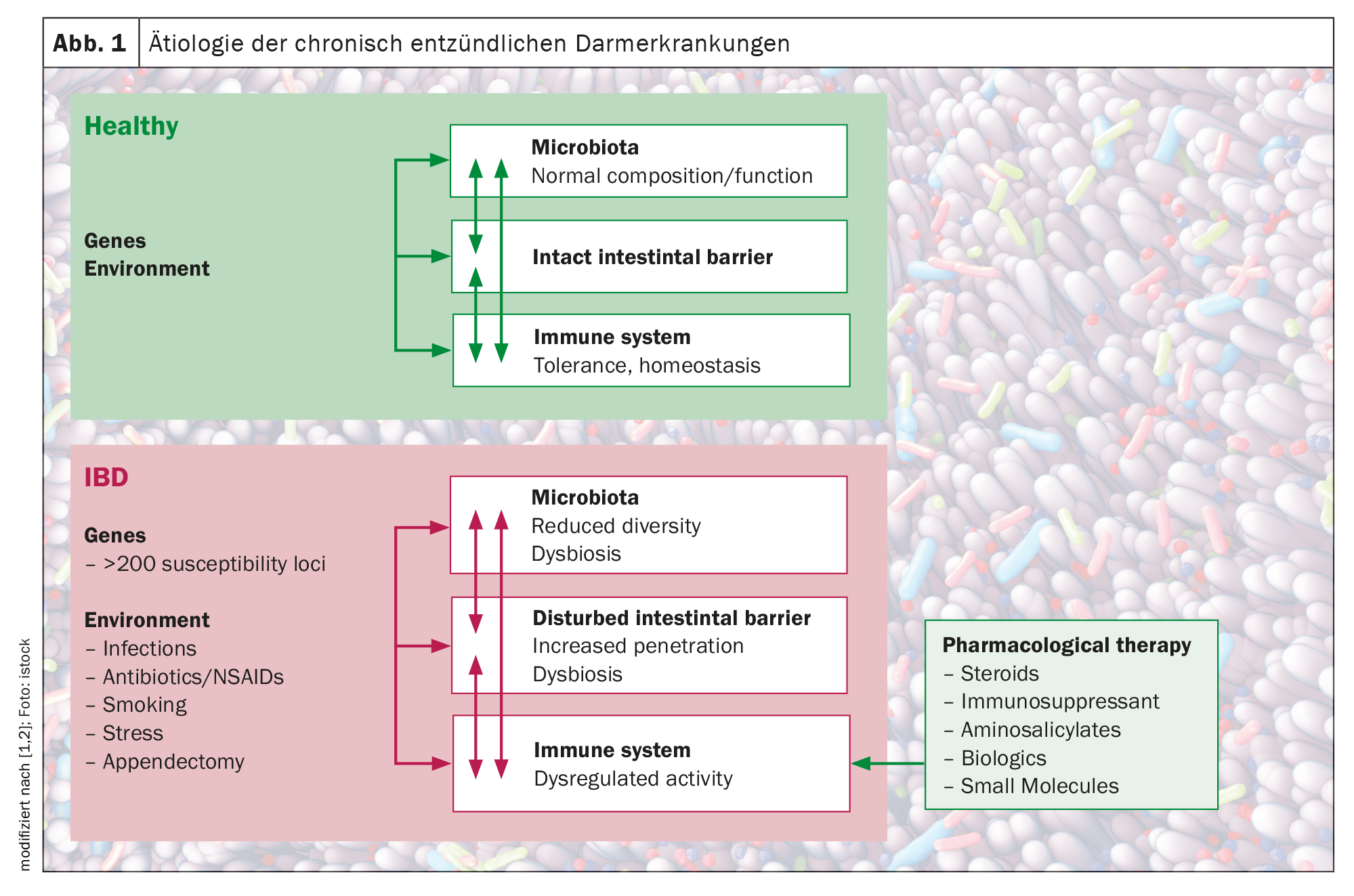
Las enfermedades inflamatorias intestinales (EII), como la enfermedad de Crohn (EC) y la colitis ulcerosa (CU), se caracterizan por la activación incontrolada de las células inmunitarias del intestino en un individuo genéticamente susceptible. Hasta la fecha, la inmunopatología de la EII no puede explicarse por completo. Sin embargo, se han investigado continuamente los componentes individuales que contribuyen a la progresión de este proceso inflamatorio crónico, incluidos los factores ambientales, los procesos alterados de migración de las células inmunitarias, así como los factores genéticos, microbianos e inmunológicos. Tras el contacto del organismo con un antígeno, se produce la activación de las células presentadoras de antígenos (CPA) (respuesta inflamatoria frente a respuesta tolerogénica). Las APC pueden producir mediadores como la interleucina 12, que conducen a la activación, proliferación e impronta de células T con un fenotipo intestinal mediante la regulación al alza de moléculas de adhesión específicas. Una barrera intestinal defectuosa y la disbiosis microbiana conducen a una acumulación y activación local de las células inmunitarias, lo que da lugar a un bucle de citoquinas proinflamatorias que anula las señales antiinflamatorias y provoca una inflamación intestinal crónica. Los estudios de asociación genética han identificado más de 250 genes de susceptibilidad para la enfermedad inflamatoria intestinal, revelando aspectos fundamentales de la biología molecular de la enfermedad, incluido el papel de la autofagia y la señalización y el desarrollo de las células Th17.
Además de las influencias genéticas, incluidos los polimorfismos genéticos del huésped en una serie de genes implicados en el reconocimiento y el procesamiento microbianos, también se ha descubierto que factores ambientales como el estilo de vida, la dieta y los medicamentos afectan al equilibrio, a menudo a través de su influencia en la composición de la microbiota intestinal. Actualmente está ampliamente aceptado que la EII es el resultado de una “tormenta perfecta” de interacciones entre una microbiota disbiótica, un sistema inmunitario anormal y las influencias ambientales en un huésped susceptible, explica el Prof. Dr. Michael Scharl, Director Adjunto de EII. Director de la Clínica de Investigación y Docencia de Gastroenterología y Hepatología del Hospital Universitario de Zúrich (Fig. 1) [1,2].
Visión general de la estructura y función del sistema inmunitario asociado al intestino
Esta alteración de la barrera permite la translocación de los antígenos bacterianos presentes en los alimentos y en determinadas regiones desde la luz intestinal hasta la pared intestinal, donde se encuentran con el mayor conjunto de células inmunitarias del cuerpo humano: el sistema inmunitario musocal, prosiguió Scharl. Al contacto con el antígeno le sigue la activación de las células presentadoras de antígenos (APC) (respuesta inflamatoria frente a respuesta tolerogénica). Las APC pueden producir mediadores como la interleucina 12, que conducen a la activación, proliferación y diferenciación de células T con un fenotipo intestinal mediante la regulación al alza de moléculas de adhesión específicas. Tras la recirculación, estos subconjuntos de células T pueden migrar posteriormente a lo largo de gradientes quimiotácticos hacia el intestino como tejido diana, donde interactúan con moléculas expresadas por las células endoteliales e inician el proceso de extravasación de múltiples pasos de la homing intestinal. Una vez en el lugar de acción, las células T adaptan la composición de sus moléculas de superficie a su entorno, lo que provoca su retención en el tejido o, si no se activan, su reciclaje en la sangre y la linfa. Si la activación local de las células T por la presentación de antígenos tiene lugar en el tejido intestinal, pueden causar daños potenciales masivos en el intestino inflamado [1,2].
Las respuestas inmunitarias desreguladas causan la EII
Además de un mayor número de células T, que son especialmente prevalentes en los pacientes con EC, tanto los pacientes con EC como los pacientes con CU muestran un mayor número de las denominadas células T auxiliares de tipo 17 (células Th17), que producen la citocina característica (interleucina 17A). Además, los pacientes con CU también tienen un mayor número de células inmunitarias de tipo 2 (células Th2), que producen interleucina 5, por ejemplo. Las citocinas típicas de las células Th2 son la IL-4 y la IL-13. Se trata de ráfagas de células inmunitarias proinflamatorias activadas, y estas respuestas inmunitarias proinflamatorias están contrarreguladas por respuestas inmunitarias antiinflamatorias mediadas, por ejemplo, por células T reguladoras (IL-10 y TGF-beta) o células Th1. Estas células también pueden ser inmunopatógenas y presentar las siguientes citocinas típicas: IFN-gamma, TNF-alfa.
En concreto, el desequilibrio entre las citocinas proinflamatorias y antiinflamatorias que se produce en la EII dificulta la resolución de la inflamación y, por el contrario, conduce a la persistencia de la enfermedad y a la destrucción del tejido. Las citoquinas desempeñan un papel central en la modulación del sistema inmunitario intestinal. Las producen los linfocitos (especialmente las células T de fenotipo Th1 y Th2), los monocitos, los macrófagos intestinales, los granulocitos, las células epiteliales, las células endoteliales y los fibroblastos. Tienen funciones proinflamatorias [interleucina-1 (IL-1), factor de necrosis tumoral (TNF-alfa), IL-12] o antiinflamatorias [antagonista del receptor de la interleucina-1 (IL-1ra), IL-10, factor de crecimiento transformante β (TGFβ)]. Las concentraciones mucosas y sistémicas de muchas citocinas pro y antiinflamatorias están aumentadas en la EII. Los estudios de asociación de todo el genoma han identificado varios loci de susceptibilidad para la EII que contienen genes que codifican citocinas y proteínas implicadas en la señalización de citocinas. En concreto, se ha demostrado que las mutaciones de pérdida de función en los genes que codifican la interleucina-10 (IL-10) y el receptor de la IL-10 están asociadas a la EII de aparición muy temprana [3,4].
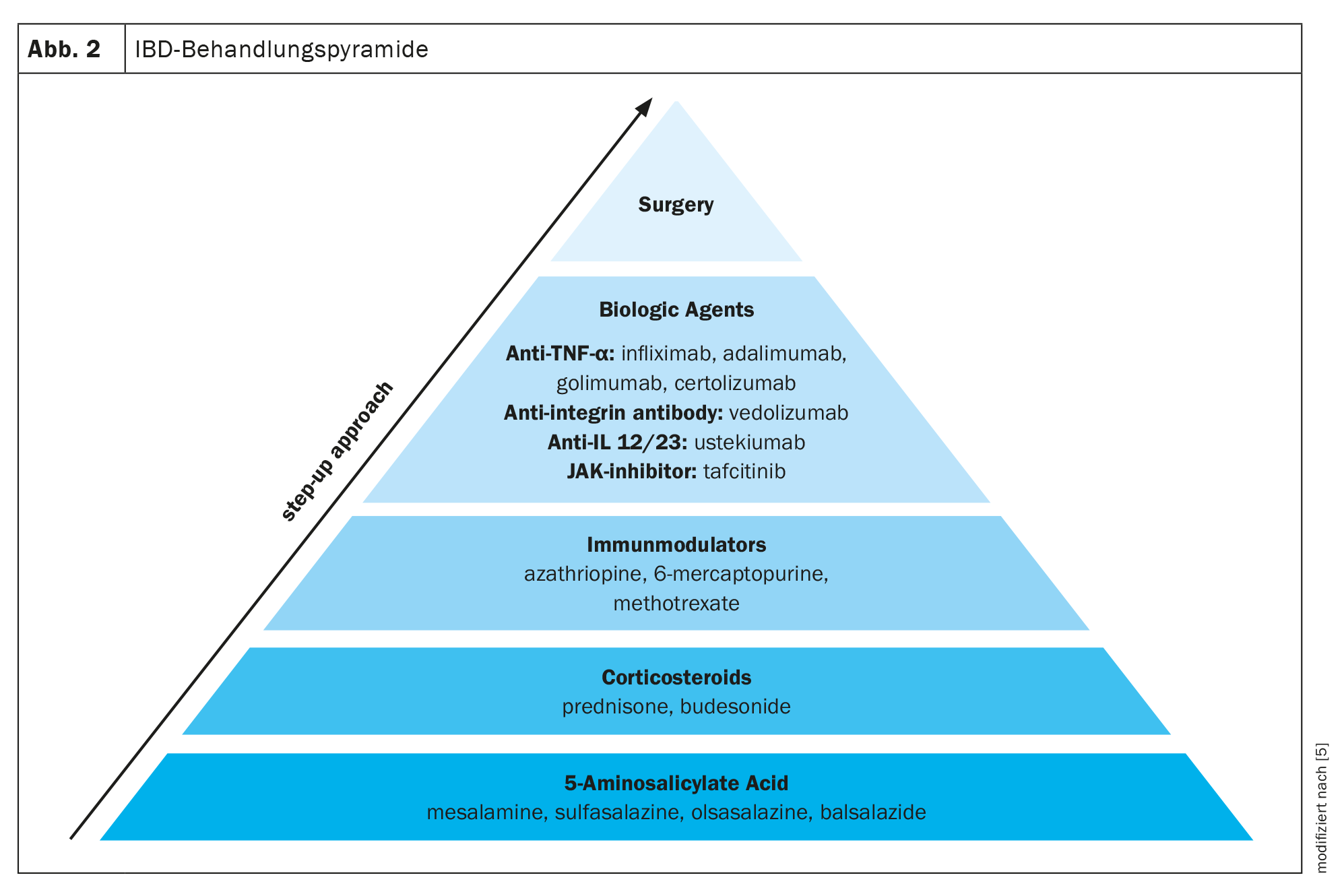
Cambios en la microbiota intestinal debidos a la terapéutica
Existen varios medicamentos para el tratamiento de la EII. A menudo se utiliza un enfoque de tratamiento por etapas, pasando de medicamentos menos específicos, como el ácido 5-aminosalicílico, a medicamentos más potentes, como los corticosteroides, los inmunomoduladores y los biológicos, en función de la gravedad de la EII. Además de la medicación, la única otra opción es la cirugía (Fig. 2) [5].
Un examen detallado de los posibles mecanismos de acción de las terapias inmunomoduladoras disponibles en la actualidad muestra que se dirigen a varias dianas potenciales en el sistema inmunitario de las mucosas, como las células inmunitarias como las células B, los macrófagos y las células T, así como a dianas en el ámbito del tráfico y la migración de células T. El ustekinumab, por ejemplo, bloquea la diferenciación en células Th1 proinflamatorias; el ozanimod inhibe la migración de células T proinflamatorias desde el ganglio linfático hacia los linfáticos de drenaje; el vedolizumab bloquea específicamente la migración de las células T efectoras proinflamatorias de los vasos sanguíneos a los tejidos intestinales; y los anti-TNF, los anti-IL12/23 y los inhibidores de JAK bloquean la función o la transcripción de las citocinas para romper el ciclo inflamatorio [5].
Ustekinumab: Diferenciación en células efectoras Th1 proinflamatorias
Una vista detallada de la diferenciación de las células T auxiliares en los ganglios linfáticos regionales muestra la activación de las células presentadoras de antígenos que producen interleucina 12. De este modo, se encuentran con células T ingenuas, que experimentan una mayor diferenciación en las denominadas células Th1. Estas células Th1 polarizadas poseen receptores de homing como el α4β7, que les permiten reinvadir la microbiota intestinal y expresar el receptor de la interleucina-12. El ustekinumab, que se dirige a la interleucina 12, puede suprimir esta vía de señalización e inhibir así la polarización de las células Th1. El anticuerpo monoclonal humano se une específicamente a la subunidad p40 de la IL-12/23, impidiendo que la IL-12 y la IL-23 se unan a sus complejos receptores de la superficie celular, bloqueando así las vías inflamatorias T helper (Th) 1 (IL-12) y Th17 (IL-23). El ustekinumab está aprobado en Suiza tanto para la EC como para la CU y se administra en infusión intravenosa durante la inducción a una dosis de inducción de 6 mg/kg. Tras una única infusión, se pasa a una terapia de mantenimiento con administración subcutánea, 90 mg, q12w/q8w (revisión 1) [3,4].

Ozanimod: migración de células T efectoras proinflamatorias del ganglio linfático a los vasos linfáticos drenantes
Tras el cebado de las células T, surgen células efectoras polarizadas. Estas células abandonan el ganglio linfático regional para llegar a los vasos linfáticos eferentes y al torrente sanguíneo. Se trata de un proceso activo que crea un gradiente quimiotáctico mediado, al menos en parte, por la molécula S1P. La S1P se une al receptor S1P de las células T, esta vía permite que las células abandonen el ganglio linfático. El ozanimod, un agonista del receptor de S1P, interfiere con este punto final del gradiente e impide la salida de células T del ganglio linfático regional. De este modo, las células T afectadas, altamente polarizadas, ya no pueden volver a la circulación. El agonista del receptor S1P, que se estudió previamente en pacientes con esclerosis múltiple, está aprobado en Suiza para la CU. Ozanimod se administra por vía oral en tres fases, 0,23 mg día 1–4 qd; 0,46 mg día 5–7 qd; y 0,92 mg qd a partir de entonces (Resumen 2) [3,4].

El ozanimod se considera una nueva opción para los pacientes con CU. Sin embargo, al tratarse de un agente biológico más reciente en el tratamiento de la EII, se necesitan más datos del mundo real, más allá de los ensayos clínicos, para evaluar en qué punto se integra el agonista del receptor de la S1P en la terapia de la EII, explica el Prof. Dr. Markus Neurath, director clínico del Hospital Universitario de Erlangen. En particular, los efectos secundarios cardiovasculares reales y la necesidad de un ECG, ponen de relieve la falta de experiencia en la práctica clínica rutinaria para que este fármaco se posicione finalmente en pacientes con CU, añadió Neurath.
Vedolizumab: migración de células T efectoras proinflamatorias de los vasos sanguíneos al tejido intestinal
El vedolizumab es un anticuerpo monoclonal humanizado de inmunoglobulina G1 (IgG1). Su mecanismo de acción selectivo en el intestino distingue al vedolizumab de los biológicos existentes para el tratamiento de la EII, que se basan en la inmunosupresión sistémica. El anticuerpo de inmunoglobulina G1 (IgG1) bloquea específicamente la α4β7-integrina en la superficie de la subpoblación de linfocitos activados que circulan por el torrente sanguíneo y que están predispuestos a dirigirse al tracto gastrointestinal. Este bloqueo interrumpe un mecanismo fisiopatológico esencial de la EII que suele permitir que los linfocitos se adhieran al endotelio del tracto gastrointestinal. Sin esta adhesión, los linfocitos ya no pueden migrar del torrente sanguíneo al tracto gastrointestinal inflamado, lo que hace que la inflamación localizada remita y sienta las bases para el control a largo plazo de la enfermedad. El vedolizumab no interrumpe el mecanismo de homing de las poblaciones de linfocitos hacia otros tejidos, por ejemplo hacia el sistema nervioso central (SNC), sino que actúa como un fármaco dirigido selectivamente a la pared intestinal mediante una inmunosupresión no sistémica. El anticuerpo inmunoglobulina G1 (IgG1) está aprobado para la EC y la CU y se administra por vía intravenosa (300 mg semana 0,2,6; después 300 mg q8w) o subcutánea (300 mg semana 0,2; después 108 mg q2w) (Visión general 3) [3,4,6].

Efectos proinflamatorios pleiotrópicos del TNF
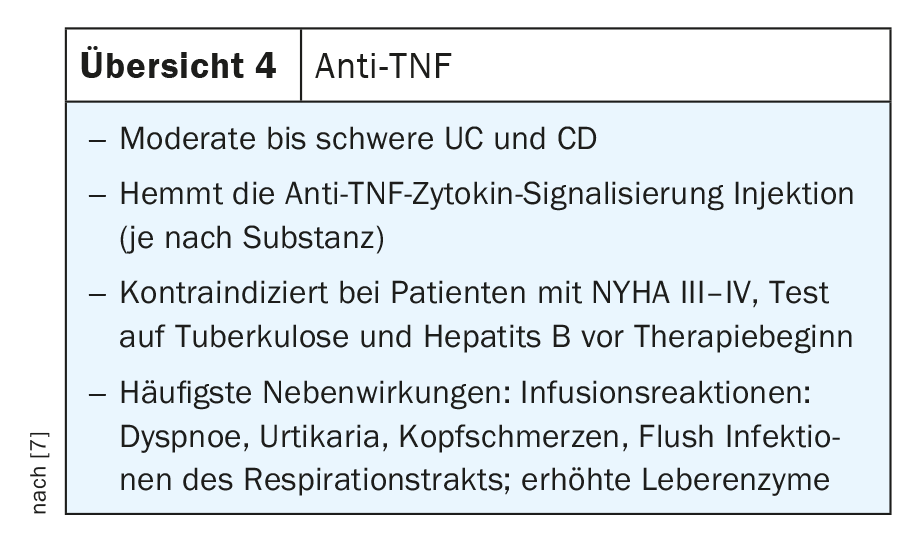
El TNF es un mediador crucial en el control de los procesos inflamatorios intestinales y se utiliza en la práctica clínica habitual desde hace más de 20 años. El TNF y sus receptores están implicados de forma crucial en la patogénesis de la EII. Por ejemplo, se hallaron niveles elevados de la forma soluble de TNFR1 y TNFR2 tanto en pacientes con EC como con CU, y su expresión se correlacionó con la actividad de la enfermedad.
Los anticuerpos anti-TNF bloquean tanto la forma precursora transmembrana (mTNF) como la forma soluble (sTNF), reduciendo así el medio proinflamatorio en el intestino al bloquear la interacción entre el TNF y el receptor del TNF, bloqueando así varios tipos proinflamatorios de células inmunitarias. Además, el TNF provoca la muerte de las células epiteliales. Así pues, los anticuerpos anti-TNF tienen varios mecanismos de acción que pueden utilizarse en la práctica clínica en pacientes con EII tanto con EC como con CU. Entre los anticuerpos anti-TNF de eficacia probada para uso clínico rutinario se encuentran el infliximab, el adalimumab, el golimumab y el certoliizumab pegol, cada uno con diferentes aplicaciones. Algunos de ellos están disponibles para terapia intravenosa, unos pocos están disponibles tanto para terapia subcutánea como intravenosa y otros sólo están disponibles para administración subcutánea (revisión 4) [7].
Según Neurath, los distintos anticuerpos anti-TNF han demostrado su eficacia en la práctica clínica y aún hoy se utilizan de forma selectiva. La vía intravenosa o subcutánea depende en cierta medida del contexto clínico. Si la actividad clínica es elevada o si el paciente está hospitalizado, la administración intravenosa es sin duda una buena forma de administrar anticuerpos anti-TNF, especialmente en pacientes con una actividad muy elevada que pierden muchos anticuerpos en las heces. Neurath añade que no es absolutamente necesario determinar las mediciones del nivel de valle ni comprobar el estado de los anticuerpos. Por lo general, esto sólo se hace en pacientes con falta de respuesta o pérdida secundaria de eficacia, o en pacientes que no logran la respuesta clínica primaria deseada. En este caso, hay varias opciones, añade Neurath: cambiar a otro agente o añadir un inmunosupresor como la azatioprina para suprimir las respuestas de las células B y los anticuerpos antifármaco. El ensayo SONIC ya ha demostrado que la terapia combinada como azatioprina más infliximab es superior a la monoterapia con sólo uno de los dos. Sin embargo, la terapia combinada debe depender de la actividad clínica. Alternativamente, el paciente puede ser monitorizado clínicamente, por ejemplo con ecografías para proteger los niveles de PCR y las actividades clínicas para determinar si el paciente está en remisión clínica. Si surgen problemas, la terapia puede entonces intensificarse, por ejemplo acortando el intervalo de infusión y aumentando la dosis del agente activo, o cambiarse a otra clase de agentes biológicos.
Actividad de las células efectoras Th17 proinflamatorias
El ustekinumab es un anticuerpo que no sólo bloquea la IL 12, sino también la interleucina 23. La IL-23 conduce a la activación y el mantenimiento de las funciones efectoras de las células Th17 proinflamatorias en el tejido intestinal. Ustekinumab reduce la actividad de las células Th17 proinflamatorias en el tejido intestinal mediante el bloqueo de la interacción Il-23/Il-23R. Basándose en estos hallazgos, actualmente se está intentando desarrollar antagonistas selectivos contra la interleucina 23. No están dirigidos a la subunidad P40, como es el caso del ustekinumab, sino a la subunidad P19, que es exclusiva de la interleucina 23 y no se encuentra en la interleucina 12. Algunos agentes para ello ya se encuentran en ensayos clínicos y tarde o temprano se utilizarán en la práctica clínica, por ejemplo el risankizumab, el mirikizumab, el guselkumab y el brazikumab. Además, algunos datos preliminares sugieren que los bloqueantes P19 también pueden ser eficaces cuando los bloqueantes P40 no han funcionado previamente (revisión 5) [3,4].

Señalización de citoquinas a través de las vías de señalización JAK/STAT
La vía de señalización de la quinasa Janus y activadora de la transcripción (JAK-STAT) desempeña un papel importante en la transmisión de señales desde los receptores de la membrana celular hasta el núcleo. Muchas citoquinas proinflamatorias inducen la transcripción de genes efectores en la célula diana mediante la activación de vías de señalización JAK/STAT específicas. La familia JAK humana consta de cuatro JAK: JAK1, JAK2, JAK3 y TYK2. El tofacitinib, un inhibidor que se dirige principalmente a las JAK 1 y JAK 3, y en menor medida a la JAK 2, reduce la transcripción de los genes efectores y de señalización proinflamatorios mediante el bloqueo de la actividad de la quinasa JAK. El tofacitinib es una pequeña molécula que actúa simultáneamente sobre varias citocinas y está disponible por vía oral. Sin embargo, hasta ahora el inhibidor sólo ha sido aprobado para la CU, pero no para la EC (resumen 6) [8–10].

Debido a su actividad inmunomoduladora y al riesgo de acontecimientos cardiovasculares y tromboembólicos, se han impuesto restricciones de uso desde que se aprobó el tofacitinib, por lo que no es el fármaco de primera elección, dijo Neurath. En particular, los pacientes de edad avanzada con enfermedades cardiovasculares y un riesgo potencialmente mayor de episodios tromboembólicos deben someterse a pruebas minuciosas antes de iniciar la terapia.
En Suiza, actualmente sólo se dispone de tofacitinib para la inhibición de JAK. Sin embargo, debido a la diversidad de estudios, cada vez se aprueban más inhibidores de JAK o se encuentran en ensayos clínicos, por lo que tarde o temprano se dispondrá de toda una gama de inhibidores de JAK para la práctica clínica. Aquí, los cambios más pequeños en la afinidad de las moléculas supondrán una gran diferencia clínica. Según Neurath, podría haber diferencias relevantes entre los distintos inhibidores de JAK-1, por ejemplo, pero esto aún debe investigarse. La patogénesis deja claro que estos agentes interfieren en la activación de las células inmunitarias. Será interesante comparar la eficacia, pero sobre todo el perfil de seguridad, añade Neurath. Esto se debe a que la seguridad, en particular, desempeña un papel decisivo para la rutina clínica, pero también para los pacientes.
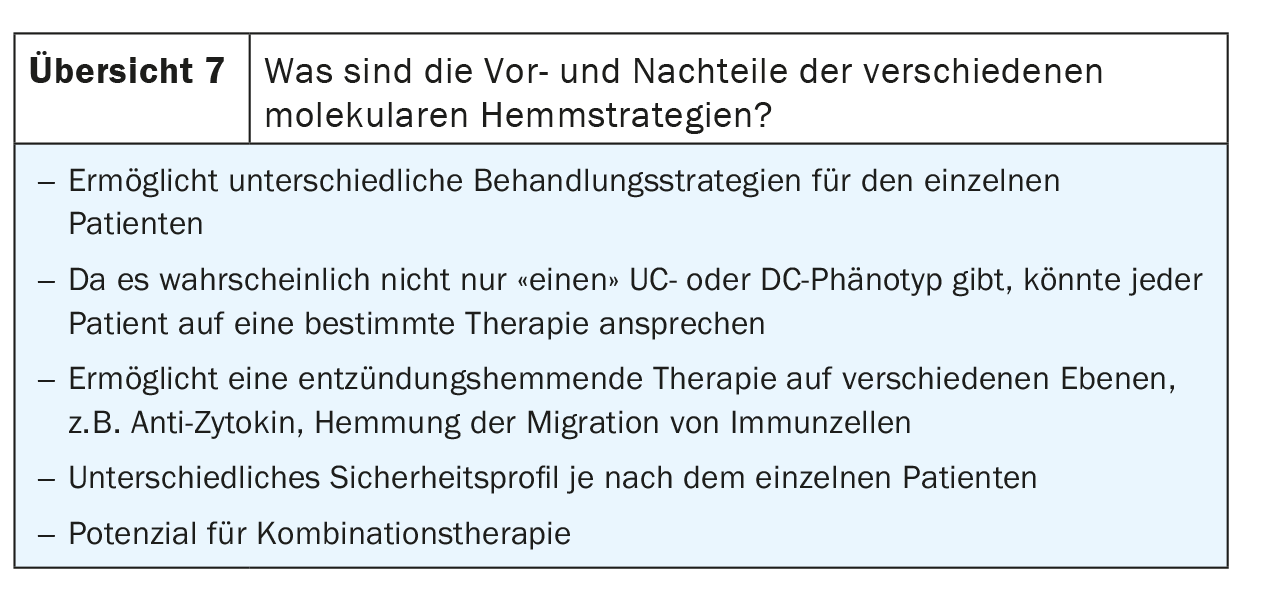
Opciones futuras y terapias combinadas para la EII
Según Neurath, se dispone de los primeros datos de un estudio de combinación con bloqueantes P19 más anticuerpos anti-TNF y se están realizando más estudios [11]. Esencialmente, aquí se utilizó una terapia combinada para inducir la remisión, por lo que no se trata de una terapia combinada de por vida. Sin embargo, podría ser una opción para los pacientes difíciles de tratar. El vedolizimab en particular parece ser un socio atractivo para las terapias combinadas porque tiene un mecanismo de acción molecular muy diferente en comparación con otros agentes, por lo que es fácil al menos postular que podría haber sinergias, añadió Neurath. Además, es un agente muy seguro que tiene un buen perfil de seguridad, lo que lo convierte en una línea de base ideal para cualquier enfoque combinado. Sin embargo, Neurath no ve mucho potencial en la combinación de inhibidores de JAK con agentes biológicos.
Mensajes para llevar a casa
- Las terapias inmunomoduladoras se dirigen a diferentes dianas potenciales en el sistema inmunitario de las mucosas.
- El ustekinumab bloquea la diferenciación en células Th1 (a través de la IL-12), así como la citocina IL-23.
- El ozanimod inhibe la migración de las células T proinflamatorias del ganglio linfático a los vasos linfáticos que lo drenan.
- El vedolizumab bloquea específicamente la migración de las células T efectoras proinflamatorias desde los vasos sanguíneos al tejido intestinal.
- Los anti-TNF, los anti-IL12/23 y los inhibidores JAK bloquean la función de las citocinas para detener la inflamación.

Literatura:
- Neurath MF: Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol 2019; doi: 10.1038/s41590-019-0415-0.
- de Lange KM, et al.: Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 2017; doi: 10.1038/ng.3760.
- Neurath MF: Cytokines in inflammatory bowel disease. Nat Rev Immunol 2014; doi: 10.1038/nri3661.
- Neurath MF: Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol 2017; doi: 10.1038/nrgastro.2016.208.
- Wu N, et al.: Inflammatory bowel disease and the gut microbiota. Proc Nutr Soc 2021; doi: 10.1017/S002966512100197X.
- Denucci CC, et al.: Integrin function in T-cell homing to lymphoid and nonlymphoid sites: getting there and staying there. Crit Rev Immunol 2009; doi: 10.1615/critrevimmunol.v29.i2.10.
- Billmeier U, et al.: Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J Gastroenterol 2016; doi: 10.3748/wjg.v22.i42.9300.
- Vetter M, Neurath M: Emerging oral targeted therapies in inflammatory bowel diseases: opportunities and challenges. Therap Adv Gastroenterol 2017; doi: 10.1177/1756283X17727388.
- Seif F, et al.: The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal 2017; doi: 10.1186/s12964-017-0177-y.
- Danese S, et al.: JAK selectivity for inflammatory bowel disease treatment: does it clinically matter? Gut 2019; doi: 10.1136/gutjnl-2019-318448.
- Feagan BG, et al.: Guselkumab plus golimumab combination therapy versus guselkumab or golimumab monotherapy in patients with ulcerative colitis (VEGA): a randomised, double-blind, controlled, phase 2, proof-of-concept trial. Published :February 01, 2023. DOI: https://doi.org/10.1016/S2468-1253(22)00427-7
HAUSARZT PRAXIS 2023: 18(6): 6–11