
La terapia sigue centrándose en las opciones de tratamiento sintomático y en evitar los factores de provocación. Las opciones terapéuticas moleculares, potencialmente también curativas, para la epidermólisis bullosa se encuentran aún en fase de investigación. En el trabajo de diagnóstico de esta enfermedad autoinmune heterogénea desde el punto de vista genético y fenotípico, hay que tener en cuenta varias cosas.
La epidermólisis bullosa (EB) es un grupo de genodermatosis heterogéneas desde el punto de vista genético y fenotípico que se caracterizan por una excesiva fragilidad mecánica de los tejidos epitelizados. Con una prevalencia de unos 500.000 casos en todo el mundo, la EB es una enfermedad rara [1,2]. Hasta la fecha, se han descrito mutaciones en más de 20 genes que codifican componentes implicados en el ensamblaje de los filamentos de queratina del citoesqueleto, las moléculas de adhesión, los desmosomas, los hemidesmosomas y las fibrillas de anclaje de los epitelios. (Fig. 1). En consecuencia, se deteriora la integridad estructural y funcional de la adhesión intraepidérmica y dermoepidérmica de la piel y las mucosas, lo que afecta a la función de barrera, la interacción célula-célula y célula-matriz, la proliferación, la regeneración tisular y los procesos de diferenciación. [3–5]. El espectro combinatorio del tipo, la homocigosidad o heterocigosidad, el número (herencia monogenética o digenética) y la localización de la mutación en el segmento génico respectivo, así como la perturbación cuantitativa (ausencia o reducción) y cualitativa (pérdida gradual de la función) resultante de la expresión de la proteína, provocan considerables variaciones genéticas y, en consecuencia, fenotípicas de la EB. Además del defecto genético primario, los factores epigenéticos y bioquímicos secundarios (por ejemplo, la inducción de cascadas inflamatorias crónicas y la remodelación tisular), así como los factores ambientales, también influyen en la gravedad clínica [6,7].
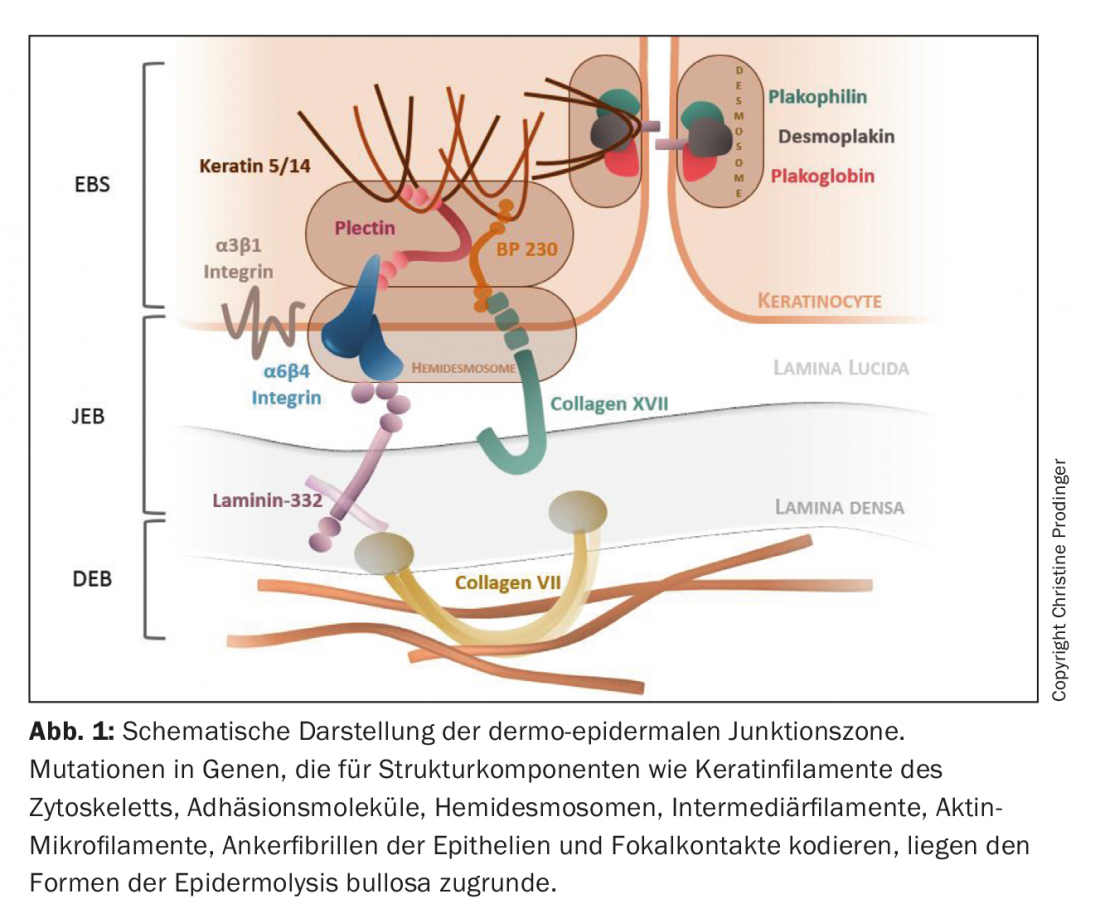
Dado que los genes índice afectados no sólo se expresan en la piel y las mucosas próximas a la piel, sino también en otros epitelios (tractos respiratorio, urogenital y gastrointestinal) y en el tejido mesenquimal (músculos esqueléticos), también son posibles las manifestaciones extracutáneas primarias. Así, la EB puede convertirse en una enfermedad sistémica con una morbilidad y mortalidad significativas [8].
Clasificación fisiopatológica
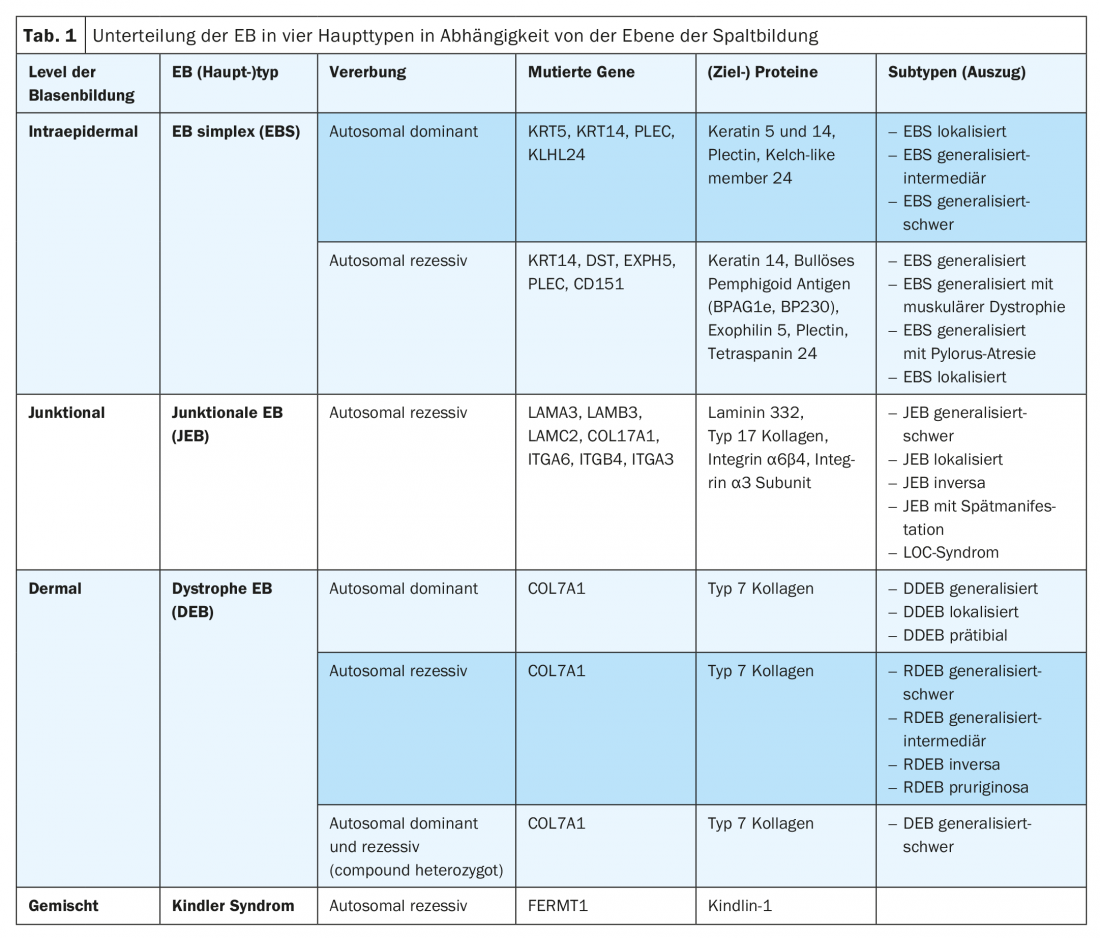
El síntoma principal de la EB es el aumento a excesiva fragilidad de la piel y las mucosas frente a la tensión mecánica con la formación de ampollas, erosiones, ulceraciones, costras o cicatrices, dependiendo del subtipo. Según el nivel de escisión como función y consecuencia de la mutación subyacente, la EB se divide en cuatro tipos principales (Tab. 1) [9]. Debido a la creciente disponibilidad de métodos modernos de diagnóstico molecular (por ejemplo, la “secuenciación de próxima generación”), también se describen una y otra vez nuevos (sub)tipos de EB. Así, una mutación en el gen KLHL24 que codifica un componente del complejo ubiquitina ligasa podría asignarse a una variante autosómica dominante de EB simplex, que da lugar a una ubiquitinación y degradación excesivas de la queratina 14 [10–12]. En un fenotipo similar al del síndrome de Kindler, también se identificó recientemente una mutación en el gen CD151, que codifica una tetraspanina de la zona de la membrana basal. Esta proteína transmembrana interactúa con las integrinas y está implicada en los procesos de crecimiento, desarrollo y motilidad celular [13]. Además, recientemente se ha descubierto una mutación en el gen PLOD3, que codifica la lisil hidroxilasa 3 y regula el procesamiento postraduccional del colágeno de tipo 7. Clínicamente, los individuos afectados muestran defectos extensos del tejido conjuntivo, contracturas articulares, malformaciones esqueléticas y retraso del crecimiento. El nivel de formación de ampollas es similar al de la EB distrófica recesiva en la sublamina densa [14].
Algoritmo de diagnóstico
Especialmente en el recién nacido con formación de ampollas, debe excluirse en primer lugar una génesis traumática, metabólica, hematológica, infecciosa, medicamentosa y autoinmune [15]. Posteriormente, se aplica un algoritmo de diagnóstico:
- Historia (familiar) y clínica
- Determinación de la contaminación microbiana (por ejemplo, frotis, PCR, serología)
- Biopsia perilesional para la evaluación histológica de posibles diagnósticos diferenciales
- Inmunofluorescencia directa paralesional para determinar el nivel de escisión y la expresión semicuantitativa de la proteína a partir de una ampolla inducida (por ejemplo, rotación de la goma de borrar de un lápiz hasta que se desarrolle eritema) y fresca (no más de 12 horas) en piel no expuesta al sol.
- Microscopía electrónica de transmisión para determinar el plano de clivaje y los defectos morfológicos
- Análisis de mutaciones (en función del caso como secuenciación del genoma completo, exoma, clúster, panel)
El (sub)tipo de EB respectivo puede determinarse en función de los hallazgos recogidos. El plano de clivaje determinado por microscopía electrónica de transmisión e inmunofluorescencia define el tipo principal de EB. El fenotipo clínico se define indicando la gravedad relativa (leve, intermedia, grave) y el patrón de distribución (localizado, generalizado); en su caso, también se indican síntomas característicos como la pseudosindactilia. Además de los hallazgos específicos recogidos en la microscopía electrónica de transmisión o la cartografía de inmunofluorescencia, se enumeran la proteína y el gen específicos afectados, el tipo de mutación o la mutación específica [9]. Los éxitos en la caracterización molecular de las bases patogenéticas de los distintos tipos de EB también permiten un asesoramiento genético, un pronóstico y un diagnóstico prenatal más precisos y son el requisito previo básico para intervenciones terapéuticas dirigidas innovadoras.
Principios terapéuticos
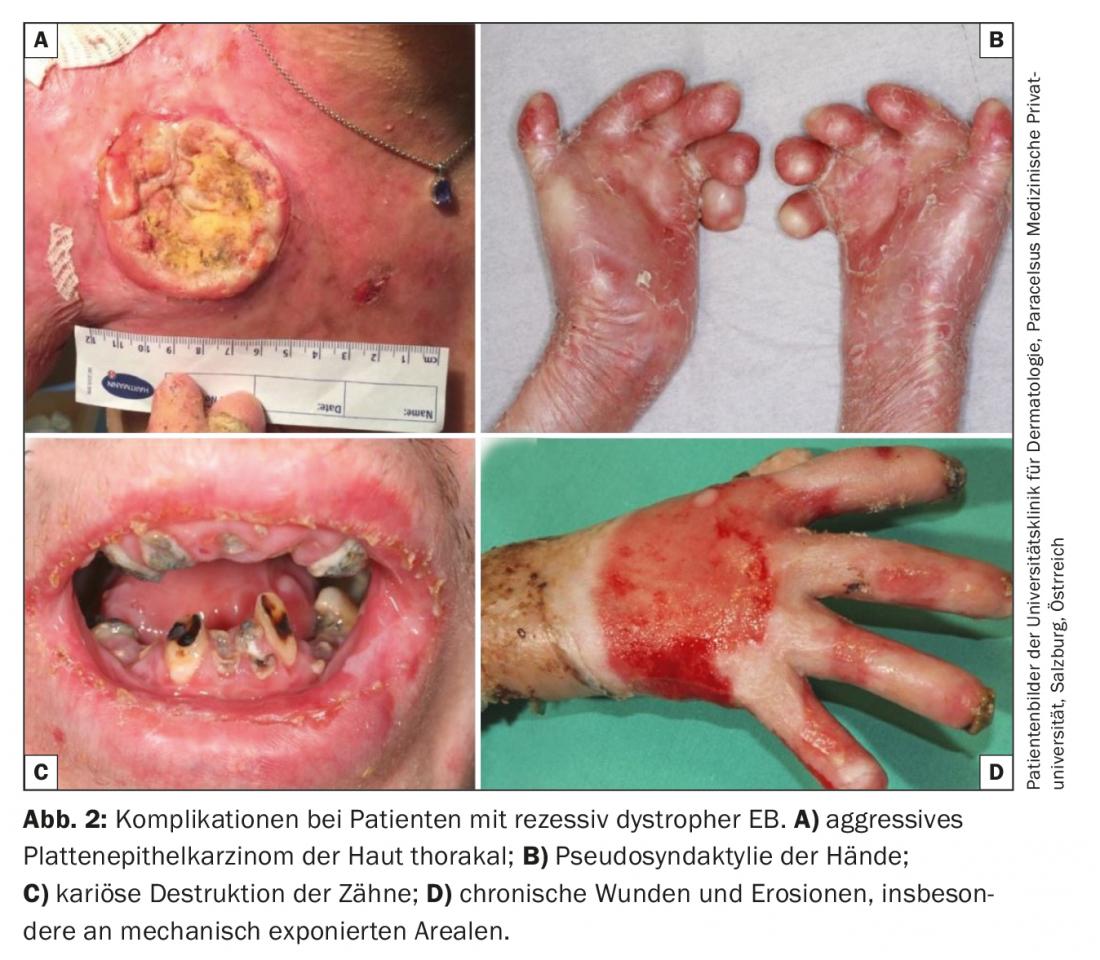
A falta de opciones de tratamiento curativo en la práctica clínica, la terapia de todas las formas de EB se centra en evitar los factores de provocación y optimizar los enfoques terapéuticos sintomáticos . Estos incluyen una terapia local adecuada al estadio de la herida, la mejora o el mantenimiento de la barrera cutánea mediante un cuidado optimizado de la piel, el alivio del dolor y el picor, la prevención y el tratamiento de las infecciones (terapia antiséptica y antibiótica intermitente), el mantenimiento de una ingesta calórica y de nutrientes adecuada y, por último, la prevención y el tratamiento de complicaciones como la anemia, la osteoporosis y el carcinoma agresivo de células escamosas. (Fig. 2) [16,17].
Las perspectivas de opciones terapéuticas moleculares, potencialmente también curativas, para la EB son, en principio, alentadoras, aunque su aplicación amplia, segura, (sosteniblemente) eficiente y practicable en la práctica clínica diaria para los pacientes con EB sea aún difícil de evaluar en la actualidad. El espectro de métodos es complejo:
Terapia génica: En 2006, se realizó por primera vez una terapia génica mediante trasplante de queratinocitos corregidos por el gen LAMB3 en un paciente con JEB autosómica recesiva (mutación en el gen LAMB3) [18]. En esta terapia de “sustitución genética “, se transfectaron células cutáneas cultivadas con un vector que contenía una copia de ADNc funcional del gen mutante causante y se trasplantaron en zonas heridas como equivalentes cutáneos epiteliales. La zona de unión dermoepidérmica se ha mantenido estructural y funcionalmente estable durante el periodo de seguimiento de 14 años hasta la fecha, sin indicios de ampollas, inflamación o reacción inmunológica al neoantígeno introducido terapéuticamente. En un estudio reciente, se cultivaron queratinocitos autólogos según el mismo principio, se aislaron células madre epidérmicas y se corrigieron genéticamente de forma específica, se expandieron y luego se trasplantaron en grandes zonas de heridas de un niño de 7 años con JEB (mutación en el gen de la laminina 332). De este modo, se pudo lograr una reepitelización del 80% de la superficie corporal. La respuesta clínica se correlacionó con la expresión persistente de laminina 332 hasta la fecha [19]. La eficacia está limitada por el número (demasiado) pequeño de células madre epidérmicas en los cultivos primarios de pacientes con EB para garantizar una corrección permanente, que se agotan debido a las heridas crónicas y al avance de la edad, así como por los aspectos de seguridad (en relación con los vectores virales y la genotoxicidad o la mutagénesis insercional inducida). En los estudios actuales se están evaluando los avances tecnológicos, que deberían mejorar el perfil de riesgo-beneficio de este método, especialmente adecuado para heridas crónicas circunscritas muy sintomáticas o con riesgo de complicaciones a largo plazo [20].
También están en desarrollo el “silenciamiento génico” para las formas autosómicas dominantes de EB (es decir, silenciar el alelo mutante con “pequeños ARN de interferencia”), así como enfoques de terapia génica tópica de fácil aplicación. En este último caso, por ejemplo, el vesículas extracelulares, que se aíslan a partir de células madre mesenquimales alogénicas y pueden transportar tanto la proteína de colágeno tipo 7 que falta como el ARNm COL7A1 a las células diana [21, 22].
Las tecnologías de edición del genoma que utilizan las propiedades de las nucleasas programables (por ejemplo, CRISPR/Cas9, TALEN, nucleasas de dedos de zinc) se basan en la modificación/corrección de secuencias genéticas mutadas. En este proceso, se induce específicamente una rotura de doble cadena en el ADN, se eliminan las secuencias mutadas y se insertan secuencias extrañas correctoras, o se corrigen bases individuales mediante mecanismos de replicación celular. Sin embargo, los problemas de seguridad, especialmente en lo que respecta a la precisión insuficiente y a los posibles sitios de unión fuera del objetivo, no justifican aún su uso (in vivo) en humanos [23,24].
Un fenómeno clínico notable que puede darse en todas las formas principales de EB es la aparición (espontánea) de zonas de piel sana sin ampollas como resultado de eventos genéticos correctores, lo que se denomina mosaicismo revertante o “terapia genética natural”. Los intentos de trasplantar queratinocitos revertientes obtenidos a partir de biopsias en sacabocados en ensayos clínicos de fase I a heridas EB afectadas tras su expansión in vitro no han cumplido hasta ahora las expectativas, debido principalmente a la pérdida progresiva de células revertientes [25,26].
Terapia celular: Los primeros resultados del trasplante de células madre de médula ósea mostraron células donantes en la piel (presumiblemente células madre pluripotentes de médula ósea reprogramadas) y una buena respuesta, en parte durante varios años, en algunos pacientes con EB distrófica recesiva, a pesar de la falta de pruebas de una concentración de colágeno tipo 7 restituida. Este éxito, cuyo mecanismo subyacente aún no está claro, se vio mermado por una mortalidad periprocedimiento significativamente mayor. Las terapias de acondicionamiento y los protocolos de trasplante revisados deberían mejorar la tolerancia [27,28].
Entre los sustratos de las terapias celulares alternativas se encuentran las células mesenquimales estromales alogénicas y las células madre mesenquimales adipogénicas aplicadas por vía intradérmica o intravenosa. Se pudo observar una mejora en la cicatrización de las heridas y una reducción de los signos inflamatorios en la piel, al menos temporalmente, lo que probablemente se deba principalmente a la inducción de procesos inmunomoduladores favorables [29]. Ya se ha demostrado un efecto clínico en estudios iniciales con fibroblastos autólogos de tipo salvaje o corregidos genéticamente inyectados por vía intradérmica, que también producen colágeno de tipo 7 además de queratinocitos [30,31].
También pueden derivarse células similares a las células madre embrionarias pluripotentes a partir de células somáticas (por ejemplo, fibroblastos o queratinocitos) mediante la transfección de tres o cuatro factores de transcripción embrionarios. El uso de las llamadas células madre pluripotentes inducidas (iPSC), que pueden diferenciarse de nuevo en varios tipos de células (por ejemplo, queratinocitos o fibroblastos), también se está investigando en EB en estudios preclínicos. El uso de células/queratinocitos revertientes para la producción de iPSC tiene un gran potencial, ya que se puede prescindir de la corrección génica y de los riesgos asociados [32,33].
Terapias proteínicas: También se intenta corregir la zona de unión dermoepidérmica defectuosa mediante la sustitución de la proteína ausente o producida defectuosamente. Mientras que los estudios con la aplicación de colágeno tipo 7 recombinante se encuentran aún en fase preclínica, la aplicación tópica de un gel que contiene un colágeno tipo 7 modificado que expresa el virus del herpes simple tipo 1, por ejemplo, ya se está probando en ensayos clínicos [34,35].
Terapias basadas en ARN/”pequeñas moléculas”: Las mutaciones sin sentido, que provocan un codón de parada a través de una mutación puntual del ADN y abortan así la traducción, son responsables de alrededor del 10% de todas las enfermedades genéticas humanas. Fármacos como los aminoglucósidos (por ejemplo, la gentamicina) o el inmunomodulador amlexanox pueden provocar una “lectura” del codón de parada (por ejemplo, de la mutación COL7A1) al unirse a los ribosomas en presencia de esta mutación, permitiendo así la producción de proteínas funcionales [36].
Las modificaciones a nivel del ARN también se consiguen mediante la “omisión de exón mediada por oligonucleótidos en antisentido” (eliminación selectiva de exones que contienen mutaciones) y el “transempalme de ARN mediado por espliceosomas” (SMaRT) (corrección de segmentos de pre-ARN mutados). Esta última tecnología ya se ha utilizado preclínicamente para corregir con éxito una mutación del gen de la plectina en la EB simple y una mutación COL7A1 en la EB distrófica recesiva, así como mutaciones autosómicas dominantes en el gen de la queratina-14 de una línea celular de EB simple [37,38].
Las llamadas “pequeñas moléculas” se utilizan como mediadoras de un efecto “modificador de la enfermedad”. Entre ellos se incluye el calcipotriol tópico, que se cree que refuerza las defensas antimicrobianas endógenas y mejora la cicatrización de las heridas al aumentar la expresión del péptido antimicrobiano catelicidina [39] y la diacereína tópica, un componente de la raíz de ruibarbo y un potente inhibidor de la proinflamatoria IL-1β, que ha demostrado reducir la formación de ampollas en pacientes con EBS según datos preliminares publicados. [40]. El inhibidor oral de la tirosina cinasa rigosertib también muestra una inhibición selectiva de las células tumorales de células escamosas de pacientes con EB distrófica recesiva in vitro, y su eficacia y seguridad se están probando en un estudio en curso. El carcinoma de células escamosas altamente agresivo como complicación de la EB crónica Las heridas son una de las principales causas de muerte, sobre todo en pacientes con EB distrófica recesiva [41]. Además del rigosertib, el anticuerpo monoclonal antireceptor PD-1 nivolumab se está sometiendo actualmente a pruebas controladas para comprobar su eficacia en el carcinoma de células escamosas localmente avanzado y metastásico en la cohorte EB (EudraCT 2016-002811-16).
Mensajes para llevarse a casa
- La epidermólisis bullosa (EB) es una enfermedad heterogénea desde el punto de vista genético y fenotípico. Los cursos graves se convierten en una enfermedad multisistémica con una pronunciada morbilidad y mortalidad. Las principales causas de muerte son las infecciones, la distrofia, el fallo orgánico y el carcinoma de células escamosas. Estos últimos aparecen precozmente y se multiplican en las heridas crónicas y muestran un curso agresivo.
- El diagnóstico se realiza en correlación con la clínica, la histología, la inmunofluorescencia y el análisis molecular. A pesar de los enfoques innovadores, en parte causales, de las estrategias de terapia molecular, la curación sigue siendo una perspectiva de futuro indirectamente imprecisa. La inmunomodulación, por su parte, es una estrategia de tratamiento prometedora.
- Los componentes epigenéticos y bioquímicos secundarios, así como los factores ambientales, que pueden inducir, por ejemplo, cascadas inflamatorias crónicas y sistémicamente eficaces, tienen una gran relevancia patogenética. Como ocurre con otras enfermedades raras, ciertas características dificultan la realización de ensayos clínicos y, por tanto, la generación de pruebas de alta calidad.
Literatura:
- Fine JD: Epidermólisis bullosa hereditaria. Orphanet J Rare Dis 2010; 5: 12.
- Fine JD, et al: Epidermólisis bullosa y el riesgo de cánceres potencialmente mortales: la experiencia del Registro Nacional de EB, 1986-2006. J Am Acad Dermatol 2009; 60(2): 203-211.
- Fine JD, et al: La clasificación de la epidermólisis bullosa hereditaria (EB): Informe de la tercera reunión internacional de consenso sobre el diagnóstico y la clasificación de la EB. J Am Acad Dermatol 2008; 58(6): 931-950.
- Bruckner-Tuderman L, et al: Progresos en la investigación de la epidermólisis ampollosa: resumen de la Conferencia Internacional de Investigación DEBRA 2012. J Invest Dermatol 2013; 133(9): 2121-2126.
- Uitto J, Richard G: Progresos en la epidermólisis ampollosa: clasificación genética e implicaciones clínicas. Am J Med Genet C Semin Med Genet 2004; 131C(1): 61-74.
- Kuttner V, et al.: Remodelación global del microambiente celular debido a la pérdida de colágeno VII. Mol Syst Biol 2013; 9: 657.
- Odorisio T, et al.: Los gemelos monocigóticos discordantes para el fenotipo de epidermólisis bullosa distrófica recesiva destacan el papel de la señalización TGF-beta en la modificación de la gravedad de la enfermedad. Hum Mol Genet 2014; 23(15): 3907-3922.
- Pulkkinen L, et al: Nuevas mutaciones ITGB4 en variantes letales y no letales de epidermólisis bullosa con atresia pilórica: missense versus nonsense. Am J Hum Genet 1998; 63(5): 1376-1387.
- Fine JD, et al: Epidermólisis bullosa hereditaria: recomendaciones actualizadas sobre diagnóstico y clasificación. J Am Acad Dermatol 2014; 70(6): 1103-1126.
- He Y, et al: Mutaciones monoalélicas en el codón de iniciación de la traducción de KLHL24 causan fragilidad cutánea. Am J Hum Genet 2016; 99(6): 1395-1404.
- Lee JYW, et al: Las mutaciones en KLHL24 se suman a la heterogeneidad molecular de la epidermólisis bullosa simple. J Invest Dermatol 2017; 137(6): 1378-1380.
- Lin Z, et al.: Las mutaciones estabilizadoras de la ubiquitina ligasa KLHL24 causan la pérdida de queratina 14 y la fragilidad de la piel humana. Nat Genet 2016; 48(12): 1508-1516.
- Vahidnezhad H, et al: Una mutación recesiva en la tetraspanina CD151 causa una epidermólisis bullosa similar al síndrome de Kindler con manifestaciones multisistémicas, incluida la nefropatía. Matrix Biol 2018; 66: 22-33.
- Salo AM, et al: Un trastorno del tejido conjuntivo causado por mutaciones del gen de la lisil hidroxilasa 3. Am J Hum Genet 2008; 83(4): 495-503.
- Nischler E, et al: Errores diagnósticos en recién nacidos y bebés con ampollas y erosiones. Dermatol Res Pract 2009; 2009: 320403.
- El Hachem M, et al: Recomendaciones de consenso multicéntricas para el cuidado de la piel en la epidermólisis bullosa hereditaria. Orphanet J Rare Dis 2014; 9: 76.
- Dănescu S, et al: Correlación entre la gravedad de la enfermedad y la calidad de vida en pacientes con epidermólisis bullosa. J Eur Acad Dermatol Venereol 2018. doi: 10.1111/jdv.15371. [Epub ahead of print]
- Mavilio F, et al: Corrección de la epidermólisis bullosa juntural mediante el trasplante de células madre epidérmicas modificadas genéticamente. Nat Med 2006; 12(12): 1397-1402.
- Hirsch T, et al.: Regeneración de toda la epidermis humana mediante células madre transgénicas. Nature, 2017; 551(7680): 327-332.
- Siprashvili Z, et al: Seguridad y resultados de las heridas tras injertos epidérmicos autólogos genéticamente corregidos en pacientes con epidermólisis bullosa distrófica recesiva. JAMA 2016; 316(17): 1808-1817.
- Rosa J, et al: Sistemas actuales de administración no viral de siARN como tratamiento prometedor de enfermedades cutáneas. Curr Pharm Des 2018; 24(23): 2644-2663.
- McBride JD, et al: Mecanismo dual de transferencia de colágeno tipo VII por vesículas extracelulares de células madre mesenquimales de médula ósea a fibroblastos de epidermólisis bullosa distrófica recesiva. Biochimie 2018; 155: 50-58.
- Hainzl S, et al: Edición de COL7A1 mediante CRISPR/Cas9 en la epidermólisis bullosa distrófica recesiva. Mol Ther 2017; 25(11): 2573-2584.
- March OP, Reichelt J, Koller U: Edición génica para enfermedades cutáneas: nucleasas de diseño como herramientas para la terapia génica de los trastornos de la fragilidad cutánea. Exp Physiol 2018; 103(4): 449-455.
- Gostynski A, Pasmooij AM, Jonkman MF: Trasplante terapéutico con éxito de piel revertante en epidermólisis bullosa. J Am Acad Dermatol 2014; 70(1): 98-101.
- van den Akker PC, et al.: Una “célula revertante tardía” explica la alta frecuencia de mosaicismo revertante en la epidermólisis bullosa. PLoS One 2018; 13(2): p. e0192994.
- Ebens CL, et al: El trasplante de médula ósea con ciclofosfamida postrasplante para la epidermólisis bullosa distrófica recesiva amplía el conjunto de donantes emparentados y permite la tolerancia de los injertos celulares no hematopoyéticos. Br J Dermatol 2019. doi: 10.1111/bjd.17858. [Epub ahead of print]
- Vanden Oever M, et al: Del revés: medicina regenerativa para la epidermólisis bullosa distrófica recesiva. Pediatr Res 2018; 83(1-2): 318-324.
- Ganier C, et al: La inyección intradérmica de células estromales mesenquimales de médula ósea corrige la epidermólisis bullosa distrófica recesiva en un modelo de xenoinjerto. J Invest Dermatol 2018; 138(11): 2483-2486.
- Petrof G, et al: La terapia celular con fibroblastos mejora la cicatrización inicial en heridas de epidermólisis bullosa distrófica recesiva: resultados de un ensayo aleatorizado controlado con vehículo. Br J Dermatol 2013; 169(5): 1025-1033.
- Venugopal SS, et al: Ensayo aleatorizado de fase II controlado con vehículo de fibroblastos alogénicos intradérmicos para la epidermólisis bullosa distrófica recesiva. J Am Acad Dermatol 2013; 69(6): 898-908.e7. doi: 10.1016/j.jaad.2013.08.014
- Itoh M, Kiuru M, Cairo MS, Christiano AM.: Generación de queratinocitos a partir de células madre pluripotentes inducidas por epidermólisis bullosa distrófica normal y recesiva. Proc Natl Acad Sci U S A 2011; 108(21): 8797-8802.
- Nakayama C, et al: El desarrollo de células madre/estromales mesenquimales derivadas de células madre pluripotentes inducidas a partir de queratinocitos epidérmicos humanos normales y de RDEB. J Dermatol Sci 2018; 91(3): 301-310.
- South AP, Uitto J: Terapia de sustitución del colágeno de tipo VII en la epidermólisis bullosa distrófica recesiva: ¿cuánto, con qué frecuencia? J Invest Dermatol 2016; 136(6): 1079-1081.
- NIH: Terapia génica tópica con bercolageno Telserpavec (KB103) para restaurar el colágeno VII funcional para el tratamiento de la epidermólisis bullosa distrófica (GEM-1), https://clinicaltrials.gov/ct2/show/NCT03536143, última consulta: 25 de marzo de 2019.
- Lincoln V, et al: La gentamicina induce la lectura de la mutación sin sentido LAMB3 y restaura la laminina 332 funcional en la epidermólisis bullosa juncional. Proc Natl Acad Sci U S A, 2018; 115(28): E6536-E6545.
- Pekín P, et al: A Gene Gun-mediated Nonviral RNA trans-splicing Strategy for Col7a1 Repair. Mol Ther Nucleic Acids 2016. 5:e287. doi: 10.1038/mtna.2016.3.
- Turczynski S, et al: La omisión selectiva de exón restaura la expresión del colágeno tipo VII y la formación de fibrillas de anclaje en un modelo in vivo de RDEB. J Invest Dermatol 2016; 136(12): 2387-2395.
- Guttmann-Gruber C, et al.: Las dosis bajas de calcipotriol pueden provocar el cierre de heridas y efectos antimicrobianos y antineoplásicos en los queratinocitos de la epidermólisis bullosa. Sci Rep 2018; 8(1): 13430.
- Wally V, et al: Desarrollo del medicamento huérfano diacereína para la epidermólisis bullosa simple: Un ensayo clínico de fase 2/3 aleatorizado, controlado con placebo y doble ciego. J Am Acad Dermatol 2018; 78(5): 892-901.e7. doi: 10.1016/j.jaad.2018.01.019.
- Atanasova VS, et al: Identificación del rigosertib para el tratamiento del carcinoma de células escamosas asociado a la epidermólisis bullosa distrófica recesiva. Clin Cancer Res, 2019. doi: 10.1158/1078–0432.CCR-18–2661
PRÁCTICA DERMATOLÓGICA 2019; 29(2): 16-20









