
Las nuevas terapias en forma de anticuerpos específicos y “pequeñas moléculas” han inaugurado una nueva era. Los productos biológicos intervienen en el proceso inflamatorio de la dermatitis atópica influyendo específicamente en las citocinas individuales. Los inhibidores de JAK, por su parte, son fármacos de moléculas pequeñas cuyo objetivo es la inhibición de la vía de señalización JAK/STAT. En el sentido de la medicina personalizada, se trata cada vez más de elegir la medicación que mejor se adapte al individuo.
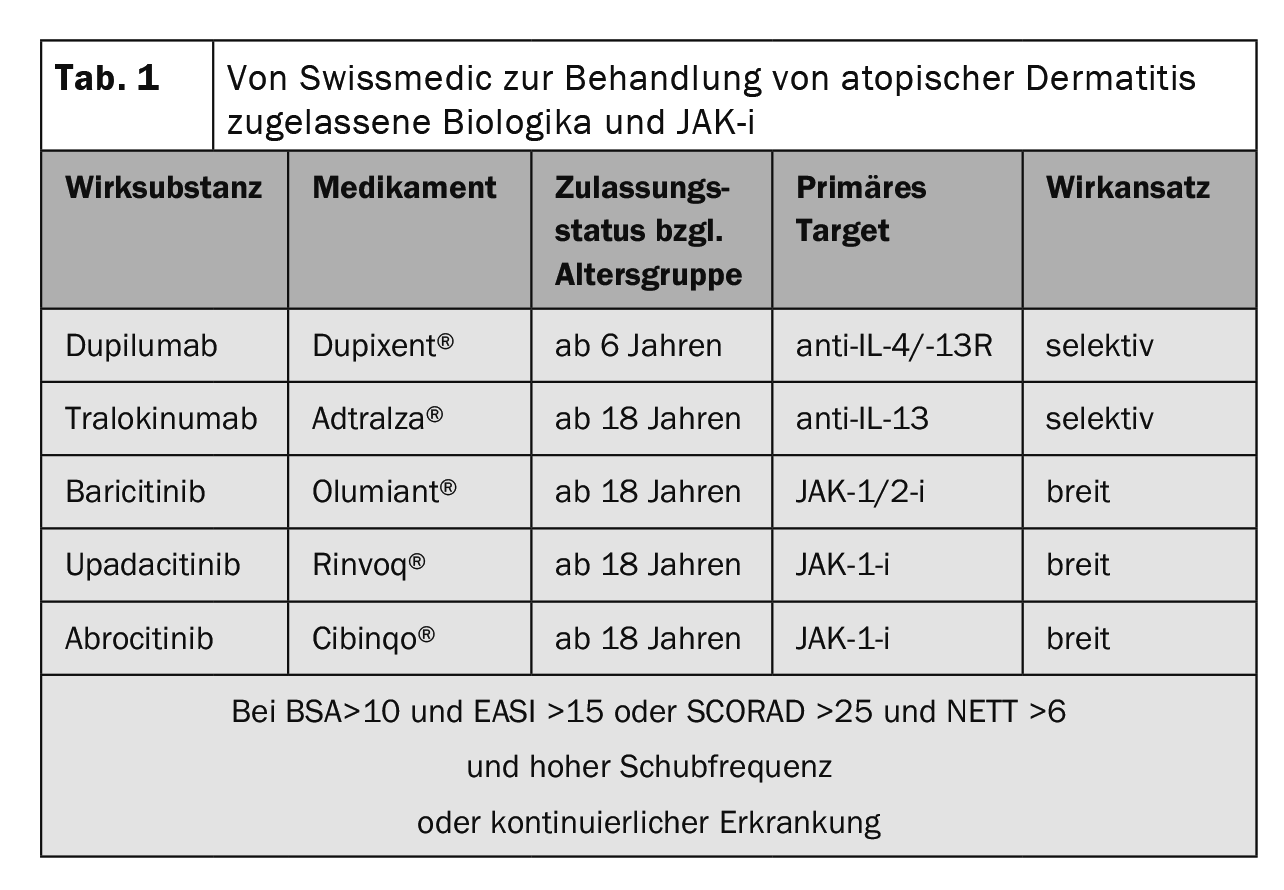
La compleja fisiopatología de la dermatitis atópica (DA) implica una predisposición genética, factores ambientales y una desregulación de la inmunidad innata y adaptativa. Las respuestas inmunitarias de tipo 2 no sólo representan el correlato celular de la inflamación cutánea en la EA, sino que también influyen significativamente en la disfunción de la barrera y la disbiosis microbiana [1,2]. La puntuación de la gravedad de la dermatitis atópica (SCORAD) puede utilizarse para definir la gravedad de la DA. Por debajo de 25 puntos el eccema atópico se clasifica como leve, entre 25 y 60 puntos como moderado y por encima de 60 como grave [3,4]. En aproximadamente una quinta parte de los casos de EA, los cursos son de moderados a graves. Los objetivos terapéuticos generales incluyen una reducción del prurito, así como una mejora de la respuesta inflamatoria y la restauración de una barrera cutánea intacta [5]. Las directrices internacionales recomiendan que el tratamiento de la EA se adapte individualmente a las distintas fases, gravedad y cronicidad según el esquema de terapia escalonada [6]. En los últimos años se han producido avances significativos, especialmente en el ámbito de las opciones de tratamiento sistémico [7]. Además de los anticuerpos monoclonales tralokinumab y dupilumab administrados como inyecciones subcutáneas, los inhibidores de la Janus quinasa (JAK) baricitinib, upadacitinib y abrocitinib están disponibles en forma de dosificación oral (Tabla 1 ).
Biológicos aprobados actualmente – IL-4 e IL-13 como citocinas clave
Las citocinas Th2 interleucina (IL)-4 e IL-13 están significativamente implicadas en las reacciones alérgicas mediadas por IgE y desempeñan un papel fisiopatológico clave en las respuestas inflamatorias y la disfunción de la barrera en la EA [10,14,25]. Un bucle de retroalimentación positiva provoca una reducción de la diferenciación de los queratinocitos y un aumento de la hiperplasia epidérmica.
El dupilumab bloquea específicamente las vías de señalización IL-4/IL-13 y es el primer anticuerpo monoclonal aprobado en el área de indicación de la dermatitis atópica. En Suiza, el dupilumab (Dupixent®) está aprobado para el tratamiento de la dermatitis atópica de moderada a grave en adultos desde 2019, seguido posteriormente de una ampliación de la indicación para adolescentes y, desde junio de 2022, el dupilumab también puede utilizarse en niños a partir de 6 años [24].
En adultos, la dosis inicial recomendada es de 600 mg de dupilumab (dos inyecciones de 300 mg) y la dosis de mantenimiento es de 300 mg cada quince días. Para niños y adolescentes de 6 a 17 años, la dosis recomendada depende del peso corporal.
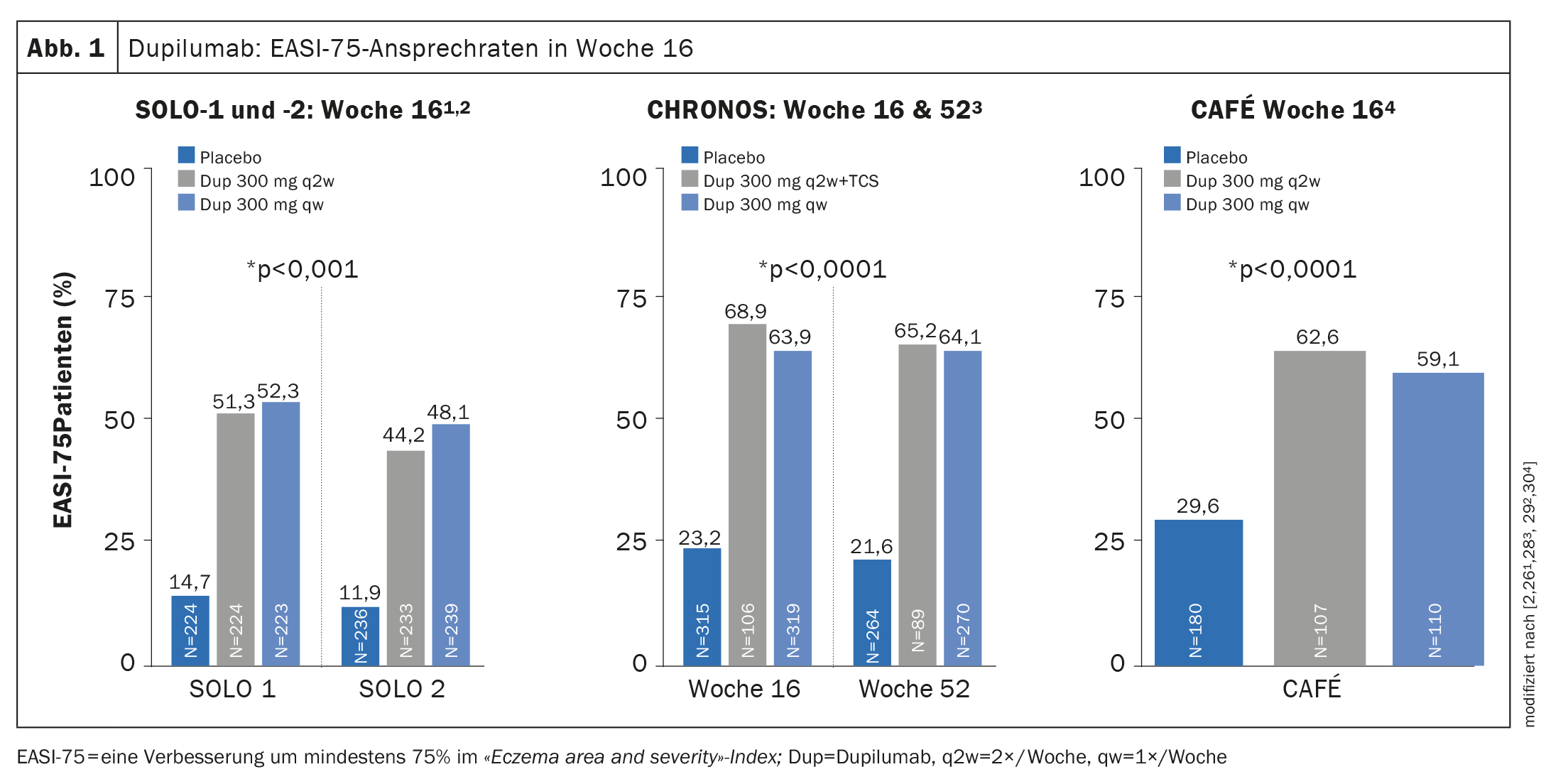
Pruebas de eficacia en ensayos de fase III: tanto en SOLO-1 y -2 (monoterapia) como en CHRONOS (terapia combinada con TCS concomitante), más del 35% de los pacientes alcanzaron una puntuación de la Evaluación Global del Investigador (IGA) de 0 ó 1 en la semana 16, lo que corresponde a una piel libre de apariencia o casi libre de apariencia [26,28]. La tasa de respuesta EASI-75 se alcanzó tanto en SOLO-1 y -2 como en CAFÉ en la semana 16 (Fig. 1) y en CHRONOS además también en la semana 52 [26,28–30]. En el estudio OLE, la tasa de respuesta IGA 0/1 alcanzó una meseta de alrededor del 60% en la semana 48, que se mantuvo hasta el punto final del estudio en la semana 76 [31,43].
Además, el dupilumab mostró un rápido alivio del prurito en una comparación con placebo, con una mejora de los valores medios mínimos cuadrados de alrededor del 20% a las dos semanas de iniciar el tratamiento [26,28–31].
La aprobación del dupilumab para adolescentes se basa en los datos clínicos del programa LIBERTY AD. La eficacia y la seguridad de la monoterapia con dupilumab a una dosis de 200 mg o 300 mg cada quince días se evaluaron en un ensayo aleatorizado, doble ciego y controlado en 251 pacientes de 12 a 17 años con EA de moderada a grave [32].
Con dupilumab, el 42% logró una respuesta EASI-75 y el 24% alcanzó un IGA 0/1. En el grupo placebo, los valores correspondientes fueron 8% y 2%. Para la población pediátrica de 6 a 11 años, la eficacia y seguridad de dupilumab en combinación con corticosteroides tópicos (TCS) se investigó en los estudios AD-1652 y AD-1434 [33].
Perfil de seguridad: Los efectos secundarios más comunes del dupilumab incluyen reacciones en el lugar de inyección y conjuntivitis, así como dolor articular, herpes oral y eosinofilia [33]. La conjuntivitis puede tratarse con éxito en la mayoría de los casos con medicamentos tópicos (colirio de ácido hialurónico o fluorometolona (0,1%) o pomada oftálmica de tacrolimus (0,03)) [34]. Se comprobó que las señales de seguridad del dupilumab en niños y adolescentes eran en general comparables a las de los adultos.
Contraindicaciones: Salvo reacciones de hipersensibilidad sistémica general muy raras, no se conocen contraindicaciones [24].
Seguimiento de la terapia y precauciones: Se recomienda actualizar el estado de vacunación de los pacientes según las recomendaciones de vacunación vigentes antes de iniciar el tratamiento con Dupilumab [24]. Las vacunas vivas y las vacunas vivas atenuadas no deben utilizarse simultáneamente con dupilumab, ya que no se ha establecido su seguridad y eficacia clínicas.
El tralokinumab es un anticuerpo monoclonal humano que neutraliza específicamente la IL-13. En Suiza, el tralokinumab (Adtralza®) está aprobado desde junio de 2022 para pacientes adultos con EA de moderada a grave cuando la terapia con medicamentos tópicos no es eficaz [24].
La dosis inicial para adultos es de 600 mg (4 inyecciones de 150 mg cada una), seguida de una dosis de mantenimiento de 300 mg (2 inyecciones de 150 mg cada una), con un intervalo de dosificación de 2 semanas cada una. A partir de la semana 16, el intervalo puede ampliarse a 4 semanas.
A diferencia del dupilumab, el tralokinumab no se une al receptor, sino a la parte soluble de la IL-13, impidiendo así la interacción con el receptor IL-13Rα1 [35,36]. La IL-13 es una citocina clave en la patogénesis de la dermatitis atópica [35]. Se ha encontrado una sobreexpresión de IL-13 tanto en la piel atópica lesional como en la no lesional, y también se ha descubierto que los niveles de IL-13 se correlacionan con la gravedad de la EA [37].
Pruebas de eficacia en ensayos de fase III: En ECZTRA-1 y -2, el 15,8% y el 22,2%, respectivamente, de los pacientes tratados cada quince días (q2w) con tralokinumab 300 mg alcanzaron un IGA 0/1 (libre de lesiones o casi libre de lesiones) en la semana 16, en comparación con sólo el 7,1% y el 10,9%, respectivamente, en el grupo de comparación con placebo [39,40]. El 25% de los participantes en el estudio tratados con tralokinumab 300 mg q2w alcanzaron el EASI-75 en la semana 16 en ECZTRA-1 y la proporción correspondiente fue del 33,2% en ECZTRA-2 (Fig. 2) [39,40].
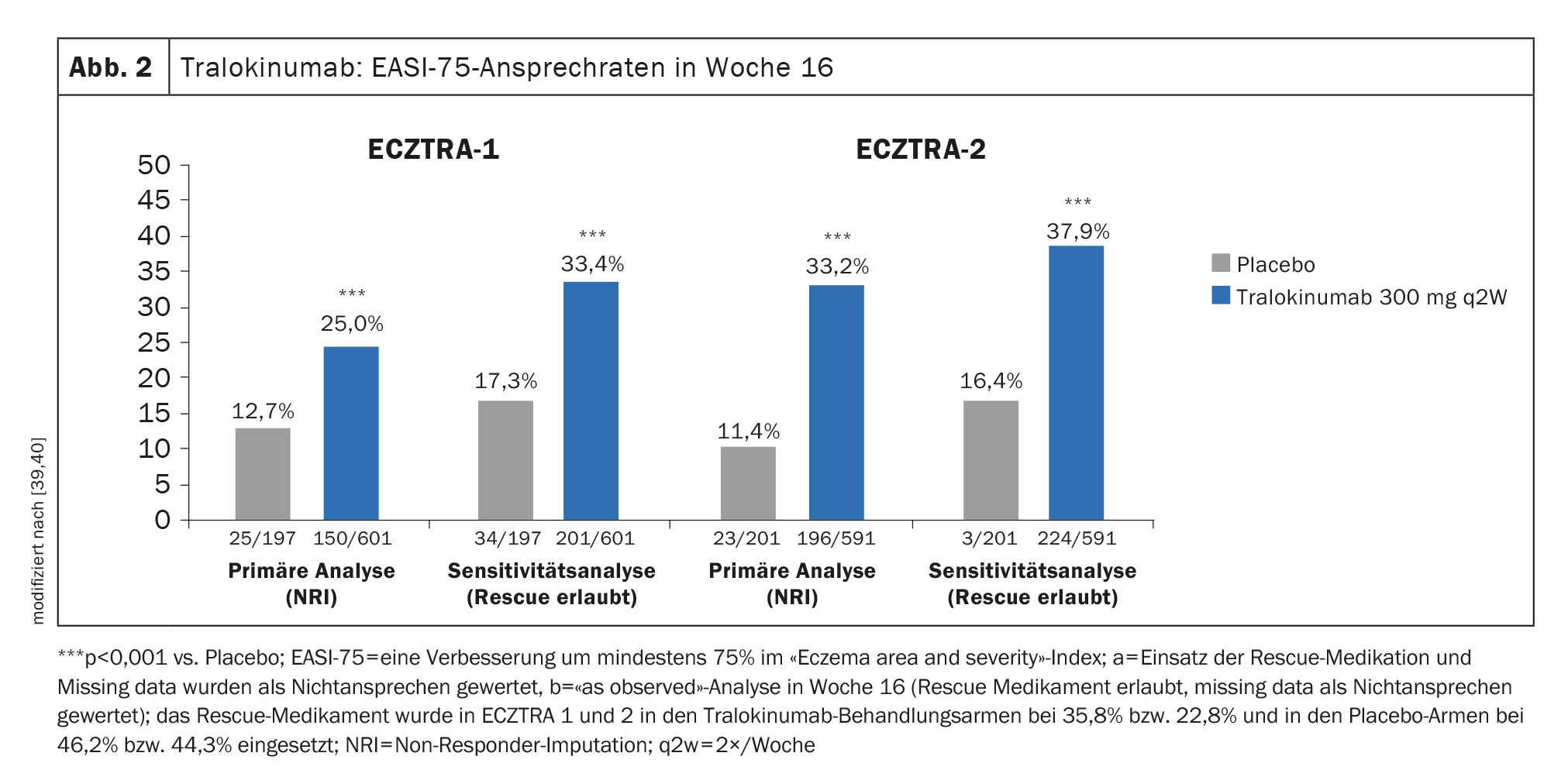
En ECZTRA-3, el tralokinumab 300 mg q2w más TCS alcanzó el EASI-75 en la semana 16, con un 56,0% de los participantes en el estudio que alcanzaron el EASI-75 en comparación con el placebo, donde esta tasa fue del 35,7%. En cuanto a la IGA 0/1, el brazo de tralokinumab también resultó ser significativamente superior en la comparación con placebo (38,9% frente a 26,2%) [37].
Perfil de seguridad: Los efectos secundarios más comunes son infección de las vías respiratorias superiores/frío y reacciones en el lugar de la inyección. La conjuntivitis se produjo con una frecuencia ligeramente menor que con dupilumab [41].
Contraindicaciones: Salvo reacciones de hipersensibilidad sistémica general muy raras, no se conocen contraindicaciones [24].
Seguimiento de la terapia y precauciones: Las vacunas vivas y las vacunas vivas atenuadas no deben administrarse simultáneamente con tralokinumab, ya que no se ha establecido su seguridad y eficacia clínicas. Los intervalos de tiempo entre una vacunación viva y el tratamiento con tralokinumab deben tomarse de las recomendaciones de vacunación actuales.
Los inhibidores JAK también van en aumento
Los inhibidores de la Janus quinasa (JAK) suprimen la respuesta intracelular de las citocinas (mecanismo JAK-STAT). El mecanismo de acción de los inhibidores de JAK consiste en utilizar “pequeñas moléculas” para bloquear las JAK de las que dependen los receptores de citocinas. Con baricitinib, upadacitinib y abrocitinib, son tres los JAK-i administrables por vía oral aprobados actualmente en Suiza para el tratamiento sistémico de la EA de moderada a grave [24]. Las citocinas patogénicamente centrales que inician su respuesta de citocinas a través de la vía JAK-STAT son la IL-4, la IL-13 y la IL-31 [44].
Lo que resulta especialmente interesante del JAK-i como opción de tratamiento es su rápido inicio de acción. El perfil de efectos secundarios de los JAK-i depende de la sustancia y de la dosis, produciéndose infecciones con más frecuencia que con los biológicos y alterándose ciertos parámetros de laboratorio durante la terapia, por lo que es necesario un seguimiento intensivo de los pacientes.
El baricitinib es un inhibidor selectivo oral de JAK1 y JAK2. A través de JAK1 se transmiten las señales de las citocinas adaptativas e innatas clave IL-4, IL-13, IL-22, IL-31, TSLP, IFN-γ e IL-6 y JAK2 es necesario para la función del receptor de IL-5, que se requiere en el reclutamiento de granulocitos eosinófilos [1].
El baricitinib (Olumiant®) está aprobado en Suiza desde febrero de 2021 para el tratamiento de pacientes adultos con dermatitis atópica de moderada a grave [24,45]. La dosis estándar es de 4 mg (1×/d p.o.). La dosis de 2 mg (1×/d p.o.) se considera en pacientes de 75 años o más o pacientes con infecciones crónicas/recurrentes o un aclaramiento de creatinina de 30–60 ml/min. Si no se evidencia ningún beneficio terapéutico del baricitinib después de 8 semanas, debe considerarse la interrupción del tratamiento.
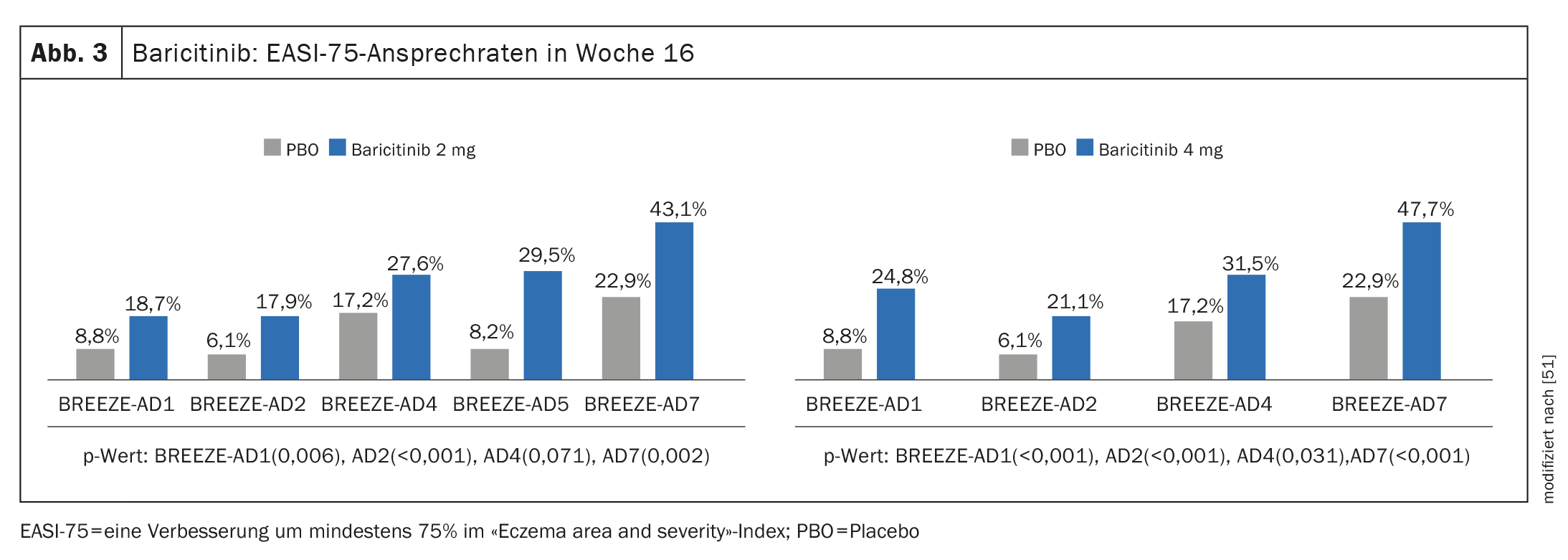
Prueba de eficacia en ensayos de fase III: Además de los ensayos BREEZE-AD1 y BREEZE-AD2, en los que la monoterapia con baricitinib produjo una reducción significativa de los síntomas, el programa de ensayos pivotales de fase III también incluyó el ensayo BREEZE-AD7 para investigar la eficacia y seguridad del baricitinib en combinación con corticosteroides tópicos (TCS) [45–47]. 329 pacientes adultos con EA de moderada a grave fueron aleatorizados en BREEZE-AD7 en una proporción 1:1:1 a baricitinib 4 mg más TCS o baricitinib 2 mg más TCS o placebo. En la semana 16, aproximadamente el 47,7% de los pacientes que recibieron baricitinib 4 mg en combinación con TCS mostraron una respuesta EASI-75, en comparación con el 22,9% del grupo placebo (p<0,001) (Fig. 3). En el brazo de 2 mg de baricitinib, los valores correspondientes fueron del 43,1% y el 22,9%, respectivamente. (Fig. 3).
Se consiguió una mejora del picor de ≥4 puntos en la escala de valoración numérica (NRS) en las primeras 16 semanas en el 44% de los pacientes tratados con baricitinib (4 mg más TCS), en comparación con el 20% con placebo más TCS (p<0,01) [48]. Se dispone de datos completos sobre la tolerabilidad del baricitinib en la EA procedentes de un total de 8 ensayos aleatorizados en los que participaron 2.531 pacientes y 2.247 pacientes-año [49].
Perfil de seguridad: Las reacciones adversas notificadas con más frecuencia durante el tratamiento con baricitinib son el aumento del colesterol LDL, las infecciones de las vías respiratorias superiores, el dolor de cabeza, el herpes simple y las infecciones de las vías urinarias [50]. La incidencia de infecciones graves con baricitinib fue similar a la del placebo.
Contraindicaciones: El baricitinib está contraindicado durante el embarazo. Las mujeres en edad fértil deben utilizar un método anticonceptivo fiable durante el uso de baricitinib y durante al menos una semana adicional después de interrumpir el tratamiento En pacientes con insuficiencia hepática grave o un aclaramiento de creatinina <30 ml/min, no se recomienda el uso de baricitinib [50].
Seguimiento del tratamiento y precauciones: Antes de iniciar el tratamiento, debe evaluarse si los pacientes presentan disfunción hepática y renal grave y antecedentes de factores de riesgo de trombosis venosa profunda o embolia pulmonar. Los niveles de lípidos (colesterol total, HDL, LDL, triglicéridos) también deben evaluarse y controlarse 12 semanas después o según la directriz sobre hiperlipidemia. Además, los pacientes deben someterse a la prueba de la tuberculosis (TB) antes de iniciar la terapia con baricitinib. El baricitinib no debe utilizarse en la tuberculosis activa, y en la tuberculosis latente no tratada previamente, debe considerarse la terapia antituberculosa antes de iniciar el tratamiento con baricitinib. Si un paciente desarrolla una infección por herpes zóster, el tratamiento con baricitinib debe interrumpirse temporalmente hasta que la infección haya desaparecido. Antes de iniciar la terapia con baricitinib debe realizarse un cribado de la hepatitis viral. Los pacientes con infección activa demostrada por hepatitis B o hepatitis C fueron excluidos de los ensayos clínicos. Se recomienda actualizar todas las inmunizaciones de acuerdo con las recomendaciones de vacunación actuales antes de la terapia con baricitinib. No se recomienda el uso de vacunas vivas atenuadas durante o inmediatamente antes del tratamiento con baricitinib.
El upadacitinib es un inhibidor de la cinasa Janus biodisponible peroralmente, selectivo y reversible que inhibe principalmente la JAK1. En Suiza, el upadacitinib (Rinvoq®) está aprobado para el tratamiento de la EA de moderada a grave en adultos desde noviembre de 2021 [52]. La dosis oral recomendada es de 15 mg de upadacitinib una vez al día.
Pruebas de eficacia en estudios de fase III: La eficacia del upadacitinib en la EA se investigó en los estudios de fase III MEASURE Up-1, MEASURE Up-2 y AD Up, con un total de 2584 pacientes incluidos en el programa de estudio. En los tres ensayos, los participantes en el estudio recibieron 15 mg o 30 mg de upadacitinib o placebo una vez al día durante 16 semanas y se demostró una mejora significativa en las puntuaciones SCORAD y EASI en la comparación con placebo. (Fig. 4). También se consiguió una mejora más rápida del aspecto de la piel y del picor [52].
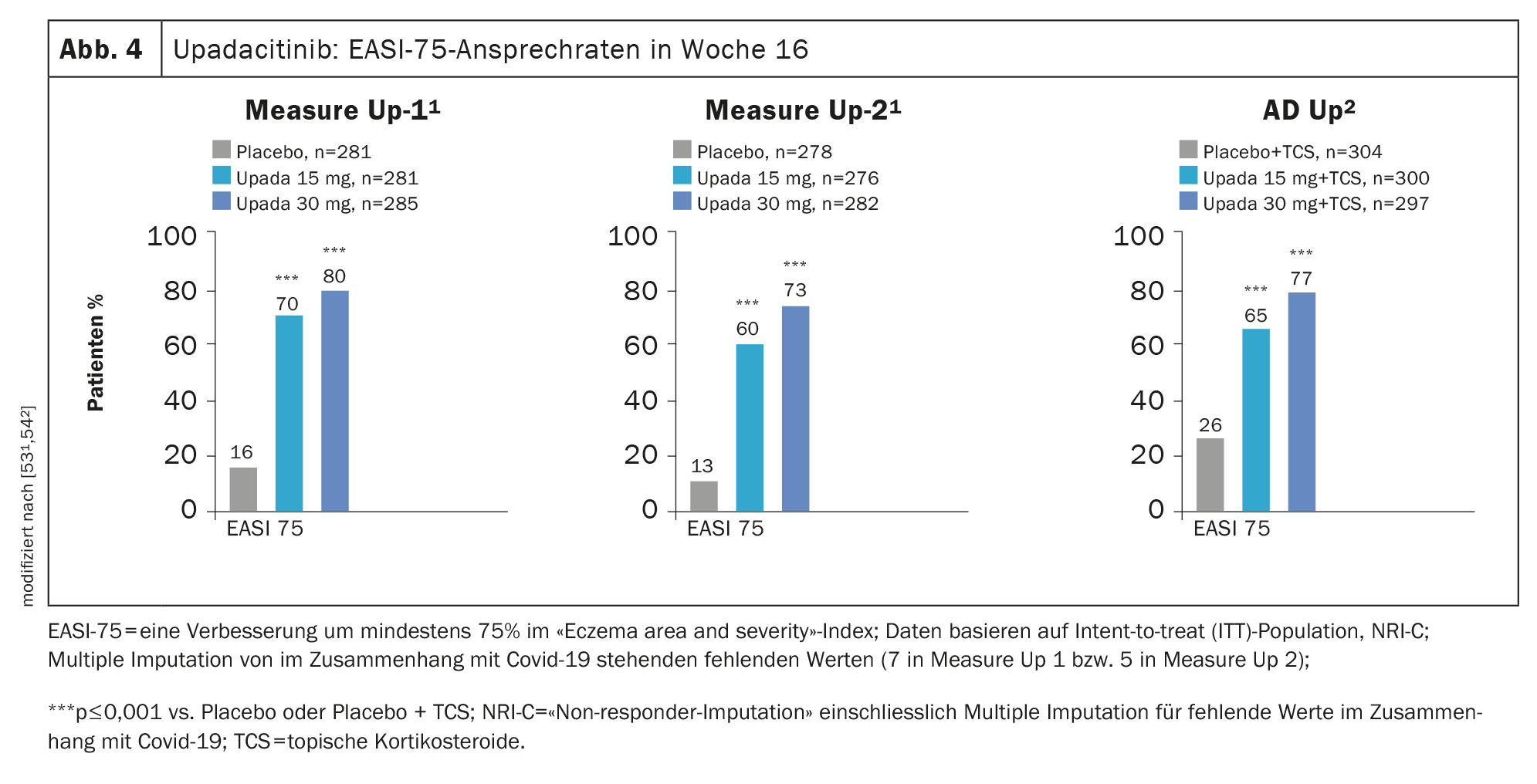
En MEASURE Up-1 y -2, se estudió el upadacitinib como monoterapia en la EA de moderada a grave en una población de sujetos que sumaba 1683 pacientes. Después de 16 semanas, más del 40% consiguió una respuesta EASI 90 y un alivio significativo del picor con upadacitinib 15 mg (1×/d), mientras que en los grupos placebo sólo se observaron ambas cosas en aproximadamente uno de cada diez pacientes [53,54]. En un análisis integrado de los ensayos Measure Up-1 y Measure Up-2, el upadacitinib mostró una respuesta EASI-90 del 62% y una mejora significativa del picor del 65% en la semana 52. [55]. Se alcanzó una respuesta EASI-75 en torno al 80% [55]. Además, el tratamiento con upadacitinib se asoció a una respuesta rápida: se observó una mejora del picor tras sólo 2 días .
Perfil de seguridad: El perfil de efectos secundarios del upadacitinib es similar al de otros inhibidores de JAK. Las más comunes son las infecciones de las vías respiratorias superiores y el acné, menos frecuentes son el herpes simple, los dolores de cabeza y la elevación de la creatina fosfoquinasa en la sangre [52].
Contraindicaciones: El upadacitinb está contraindicado durante el embarazo, las mujeres en edad fértil deben utilizar métodos anticonceptivos fiables tanto durante el tratamiento como hasta cuatro semanas después de la última dosis de upadacitinib. Se recomienda no utilizar upadacitinib en pacientes con un recuento absoluto de linfocitos inferior a 500 células/mm3, un recuento absoluto de neutrófilos inferior a 1000 células/mm3 o un nivel de hemoglobina inferior a 8 g/dl [24].
No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve o moderada. En casos de insuficiencia hepática grave (Child-Pugh C), no se recomienda el uso de upadacitinib. La insuficiencia renal no tiene una influencia clínicamente relevante en la exposición del upadacitinib [52].
Seguimiento de la terapia y precauciones: El cribado de la tuberculosis (TB) debe realizarse antes de iniciar la terapia con upadacitinib. El upadacitinib no debe utilizarse en pacientes con tuberculosis activa. En pacientes con tuberculosis latente no tratada, debe considerarse la terapia antituberculosa antes de iniciar el tratamiento con upadacitinib. Si un paciente desarrolla herpes zóster, debe considerarse la posibilidad de interrumpir el tratamiento con upadacitinib hasta que la infección haya desaparecido. Antes de iniciar la terapia con upadacitinib y durante la misma, debe realizarse un cribado de la hepatitis viral y un seguimiento para detectar una posible reactivación. Los pacientes que dieron positivo en las pruebas de anticuerpos de la hepatitis C y ARN del virus de la hepatitis C fueron excluidos de los ensayos clínicos.
Antes de iniciar la terapia con upadacitinib, se recomienda comprobar el estado de vacunación de los pacientes según las directrices de vacunación vigentes y ponerse al día en todas las vacunaciones necesarias. No se recomienda el uso de vacunas vivas atenuadas durante o inmediatamente antes del tratamiento con upadacitinib [52].
El abrocitinib es un inhibidor selectivo oral de JAK-1. En Suiza, el abrocitinib (Cibinqo®) está aprobado para el tratamiento de la dermatitis atópica en adultos desde abril de 2022 [24].
Pruebas de eficacia en estudios de fase III: La eficacia y seguridad del abrocitinib como monoterapia y en combinación con terapias de fondo activas de aplicación tópica durante un periodo de 12 a 16 semanas se investigaron en 1.616 pacientes en los estudios de fase III controlados con placebo MONO-1, MONO-2 y COMPARE. En el ensayo controlado aleatorizado REGIMEN, que también es relevante para la aprobación, se investigó el abrocitinib durante un periodo de estudio de 52 semanas en un total de 1233 participantes en el estudio [56].
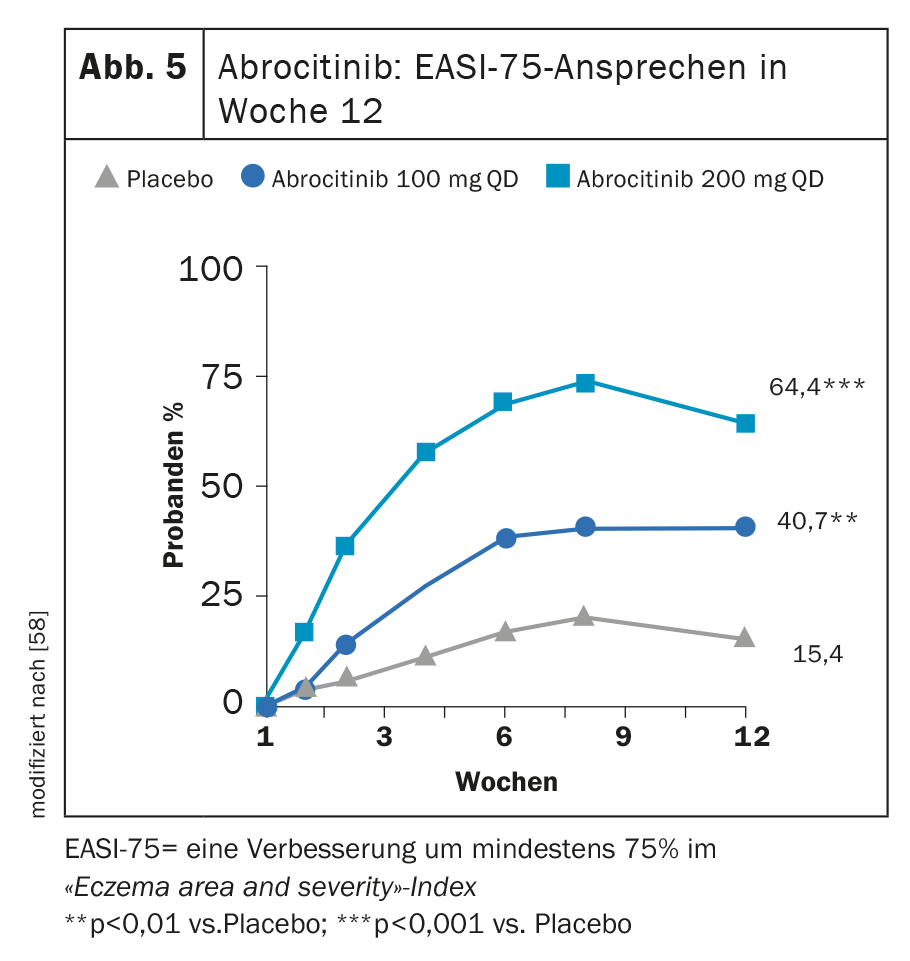
En MONO-1 y MONO-2, el tratamiento con abrocitinib 100 mg (1× d) alcanzó los dos criterios de valoración primarios IGA 0 ó 1 y/o EASI-75 en una proporción significativamente mayor en la semana 12 (Fig. 5) [24]. En combinación con el TCS, el inhibidor de JAK demostró ser significativamente superior al placebo en el ensayo COMPARE en la semana 16 con respecto a estos dos criterios de valoración. Una reducción del picor de ≥4 puntos en la NRS del prurito se manifestó con abrocitinib en comparación con placebo ya en la semana 2 en una proporción significativamente mayor de participantes en el estudio. [57]. Con la dosis alta de 200 mg, el 43,8% de los pacientes mostraron una mejoría de la AGI y el 64,6% un EASI-75 [58] tras 12 semanas en un estudio de fase IIb.
Perfil de seguridad: Los efectos secundarios notificados con más frecuencia son náuseas, dolor de cabeza, acné, herpes simple, aumento de la creatina fosfoquinasa en sangre, vómitos, mareos y dolor abdominal superior. Los efectos secundarios graves más frecuentes (0,3%) son las infecciones [56].
Contraindicaciones: El abrocitinib está contraindicado durante el embarazo. Se debe aconsejar a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento y durante un mes después de finalizar la administración peroral de upadacitinib. El abrocitinib está contraindicado en caso de insuficiencia hepática grave (Child-Pugh clase C). No es necesario ajustar la dosis en casos de insuficiencia renal leve. En caso de insuficiencia renal moderadamente grave (TFGe de 30 ml/min a <60 ml/min o TFGe <30 ml/min), deberá reducirse la dosis de acuerdo con la información sobre el producto [56].
Seguimiento de la terapia y precauciones: Los pacientes deben someterse a la prueba de la tuberculosis (TB) antes de iniciar el tratamiento con abrocitinib. El abrocitinib no debe administrarse a pacientes con tuberculosis activa. En los pacientes con tuberculosis latente no tratada, debe iniciarse la terapia preventiva para la tuberculosis latente antes de iniciar el tratamiento.
Si un paciente desarrolla una infección por herpes zóster, debe considerarse la posibilidad de interrumpir temporalmente el tratamiento con abrocitinib hasta que la infección se haya resuelto. Antes de iniciar la terapia y durante la misma, debe realizarse un cribado de la hepatitis vírica de acuerdo con las directrices clínicas. Los pacientes con infección activa demostrada por hepatitis B o hepatitis C fueron excluidos de los ensayos clínicos. Además, se recomienda poner al día todas las inmunizaciones de acuerdo con las recomendaciones de vacunación vigentes antes de iniciar el tratamiento con abrocitinib. Debe evitarse el uso de vacunas vivas atenuadas durante o inmediatamente antes del tratamiento con abrocitinib [56].
Hay otros compuestos prometedores en proyecto
Otras sustancias activas que se están investigando actualmente para su uso en la EA son [2]:
- Lebrikizumab: anticuerpo monoclonal que se une específicamente a la IL-13 que circula libremente. Además de los dos ensayos de monoterapia de fase III ADvocate 1 y 2, se está investigando la combinación de lebrikizumab con TCS en ADhere [62].
- Nemolizumab: anticuerpo monoclonal que bloquea la subunidad α del receptor de la IL-31. Los datos de los estudios realizados hasta la fecha muestran un fuerte alivio del picor en particular [63]. En un ensayo aleatorizado de 16 semanas con aleatorización 2:1, en la semana 16 el grupo de nemolizumab (n=143) mostró un cambio porcentual medio en la puntuación de la EAV de -42,8% en comparación con el -21,4% del placebo (n=72) [64].
- Tezepelumab: anticuerpo contra la TSLP (linfoproteína del estroma tímico). La TSLP es una citocina derivada de las células epiteliales que está especialmente asociada al picor en la EA [65]. En el ensayo de fase IIa ALLEVIAD, la mejora porcentual media en el pico de prurito NRS tezelumab más TCS frente a placebo en la semana 12 fue de 33,54 (DE 6,02) frente a 25,41 (DE 6,06); p=0,258) [68].
- Anticuerpo anti-Ox-40: El Ox-40 es un receptor costimulador de las células T activadas [65]. En un estudio de fase IIa aleatorizado, doble ciego y controlado con placebo, la administración del anticuerpo anti Ox-40 a intervalos de 4 semanas produjo mejoras clínicas significativas en el día 71 [70].
- Brepocitinib: inhibidor tópico de TYK2/JAK1
- Moduladores del receptor de hidrocarburos de arilo: sistema receptor citoplasmático que influye en el mecanismo inflamatorio y las moléculas de barrera.
Mensajes para llevarse a casa
- Las nuevas terapias en forma de anticuerpos específicos y las llamadas moléculas pequeñas han marcado el comienzo de una nueva era. La interleucina (IL-)4 y la IL-13 son las dos citocinas proinflamatorias más importantes en la EA. Además, hay otros mediadores inflamatorios que desempeñan un papel.
- Los productos biológicos intervienen en el proceso inflamatorio de la EA influyendo específicamente en las citocinas individuales. Los inhibidores JAK, por su parte, son fármacos de moléculas pequeñas que se dirigen a una inhibición de la vía de señalización JAK/STAT.
- Cuando una citocina se une al dominio extracelular de su receptor, las Janus quinasas activan factores de transcripción intracelulares que controlan la lectura de un gran número de genes en el núcleo celular [1].
- Las diferencias más importantes entre las terapias sistémicas modernas de gran eficacia se refieren a la forma de administración, la frecuencia de uso, el perfil de efectos secundarios y el seguimiento de la terapia. En el sentido de la medicina personalizada, se trata cada vez más de elegir el medicamento que mejor se adapte a las características de la enfermedad y a otros rasgos relacionados con la persona y el contexto.
Literatura:
- Lauffer F, Biedermann T: Dtsch Arztebl 2021; 118(24): [24]; DOI: 10.3238/PersDerma.2021.06.18.04
- “Dermatitis atópica, ¿qué hay de nuevo?”, Univ.-Prof. Dr. Paul-Gunther Sator, DermAlpin Salzburgo, 30-31.10.2021
- Deutsche Haut- und Allergiehilfe e.V, www.dha-neurodermitis.de/therapie/grundlagen-der-therapie.html,(última consulta, 06.01.2023)
- Puntuación de la gravedad de la dermatitis atópica: el índice SCORAD. Dermatología 1993; 186(1): 23-31.
- Nygaard U, Deleuran M, Vestergaard C: Dermatología 2017; 233: 333-343.
- AWMF: Directriz Neurodermatitis, Número de registro 013-027, Clasificación S2k Estado: 31.03.2015.
- Estudio bibliográfico DFP: Dermatitis atópica, www.oeadf.at/files/E-Learning/ClinicumDerma_12_032017.pdf,(última consulta: 06.01.2023)
- Leung DYM, Guttman-Yassky E: J Allergy Clin Immunol 2014; 134: 769-779.
- Kim BE, Leung DY, Boguniewicz M, Howell MDl: Clin Immunol 2008; 126: 332-337.
- Akdis CA, et al: Alergia 2020; 75(7): 1582-1605.
- Hoffjan S, Stemmer S: Arch Dermatol Res 2015; 307: 659-670.
- Darsow U, et al: J Eur Acad Dermatol Venereol 2010; 24: 317-328.
- Williams MR, Gallo RL: Curr Allergy Asthma Rep 2015; 15: 65.
- Nguyen JK, et al: Arch Dermatol Res 2020; 312(2): 81-92.
- Weidinger S, et al: Nat Rev Dis Primers 2018; 4(1): 2.
- Clark JD, et al: J Med Chem 2014; 57: 5023-5038.
- Howell MD, et al: Front Immunol 2019; 10: 2342.
- Chiesa Fuxench ZC, Ong P: Póster presentado en la AAD 2018. Póster 6236.
- Drucker AM, et al: J Invest Dermatol 2017; 137(1): 26-30.
- Ronnstad ATM, et al: JAAD 2018; 79: 448-456.
- Reich K, et al: Sociedad Internacional de Dermatitis Atópica 2021.
- Brunner PM, et al: J Invest Dermatol 2017; 137: 18-25.
- Silverberg JI: Ann Allergy Asthma Immunol 2019; 123(2): 144-151.
- Información temática, www.swissmedicinfo.ch, (última consulta: 06.01.2022)
- 25 Silverberg J: Clin Dermatol 2017; 35: 360-366.
- Simpson EL, et al: N Engl J Med 2016; 375: 2335-2348.
- Paller AS et al: J Am Acad Dermatol. 2016;75(3): 494-503.
- Blauvelt A, et al: The Lancet 2017; 389(10086): 2287-2303.
- Simpson EL, et al. Resumen de última hora, resumen D3T01.1C de la EADV 2016.
- de Bruin-Weller, M. et al: Br J Dermatol 2018; 178(5): 1083-1101.
- Deleuran M et al: J Am Acad Dermatol 2020; 82(2): 377-388.
- Simpson EL, et al: JAMA Dermatol 2020; 156(1): 44-56.
- Agencia Europea de Medicamentos: Dupilumab, Información del producto, https://ec.europa.eu/healthhttps://ec.europa.eu/health, última consulta: 06.01.2023)
- Wollenberg A, et al: J Allergy Clin Immunol Pract. 2018 Sep-Oct; 6(5): 1778-1780.e1.
- Schmid-Grendelmeier P: Dermatologie Praxis 2021; 31(4): 10-14.
- Wollenberg A, et al: JDDG 2021; 19(10): 1435-1442.
- Silverberg JI, et al: Br J Dermatol 2021; 184: 450-463.
- Petronelli M: Tralokinumab cumple los criterios de valoración de todos los estudios de fase 3, Dermatology Times, 16 de diciembre de 2019, www.dermatologytimes.com, (última consulta: 06 de enero de 2023)
- Simpson E, et al.: Presentación en AAD VMX, del 12 al 14 de junio y a la carta, 2020, EE. UU.
- Wollenberg A, et al: Br J Dermatol 2021; 184(3): 437-449.
- Agencia Europea de Medicamentos, Tralokinumab, Información del producto,https://ec.europa.eu/healthhttps://ec.europa.eu/health, última consulta: 06.01.2023)
- “Día Mundial de la Neurodermatitis el 14 de septiembre Neurodermatitis: la terapéutica sistémica mejora el tratamiento”, Berufsverband der Deutschen Dermatologen e.V. (BVVD), Berlín 03.09.2021.
- Futamura MJ: Am Acad Dermatol 2016; 74: 288-294.
- Klein B, Treudler R, Simon JC: JDDG 2022; 20(1): 19-25.
- Simpson EL, et al. Presentado en:24º Congreso Mundial de Dermatología; 10-15 de junio de 2019; Milán, Italia.
- Melo A, Carrascosa JM, Torres T: J Dermatolog Treat 2021 Ago 23: 1-10. Publicación electrónica antes de impresión.
- Wollenberg A, et al: JEADV 2021; 35(7): 1543-1552.
- Reich K, et al: JAMA Dermatol 2020; 156(12): 1333-1343.
- Silverberg JI, et al: JAMA Dermatol 2021, 157: 691-699.
- Agencia Europea de Medicamentos: Baricitinib, Información del producto, https://ec.europa.eu/health, último acceso 06.01.2023)
- 51. radi G, et al: Healthcare (Basilea) 2021 Nov 18; 9(11):1575. healthcare-09-01575-v2.pdf.
- Agencia Europea de Medicamentos: Upadacitinib, Información del producto, https://ec.europa.eu/health, último acceso 06.01.2023)
- Guttman-Yassky E, et al: Lancet 2021; 397(10290): 2151-2168.
- Reich K, et al: Lancet 2021; 397(10290): 2169-2181.
- Simpson EL, et al: Presentación en la Reunión de Recursos Esenciales de la Fundación para la Educación Dermatológica (DEF) 2021 (DERM2021), 5-8 de agosto de 2021, Las Vegas NV.
- Agencia Europea de Medicamentos: Abrocitinib, Información del producto, https://ec.europa.eu/health/, (última consulta: 06.01.2023)
- “Dermatitis atópica: la vía de señalización JAK/STAT como diana terapéutica ” https://medizinonline.com/der-jakstat-signalweg-als-therapeutische-zielstruktur,(última consulta: 16.02.2023).
- Gooderham MJ, et al: JAMA Dermatol 2019; 155(12): 1371-1379.
- Guttman-Yassky E, et al: JAMA Dermatol 2020; 156(4): 411-420.
- Moyle M, et al: Exp Dermatol 2019; 28(7): 756-768.
- 61. Loh TY, et al: J Asthma Allergy 2020; 13: 109-114.
- 62 “Eli Lilly presenta una BLA para el tratamiento de la EA con lebrikizumab”,
www.dermatologytimes.com/view/eli-lilly-submits-bla-for-lebrikizumab-ad-treatment,(última consulta: 06.01.2023) - Ruzicka T, et al: N Engl J Med 2017; 376: 826-835.
- Kabashima K, et al: N Engl J Med 2020; 383: 141-150.
- 65 Quint T, Bangert C: hautnah 2021; 20: 37-44.
- 66 Ziegler SF, et al: Adv Pharmacol 2013; 66: 129-155.
- Shikotra A, et al: J Allergy Clin Immunol 2012; 129: 104-111.e1-9.
- Simpson EL, et al: J Am Acad Dermatol 2019; 80: 1013-1021.
- Suga H, Sato S: Nuevas terapias tópicas y sistémicas en la dermatitis atópica. Inmunol Med 2019; 42(2): 84-93.
- Guttman-Yassky E, et al: J Allergy Clin Immunol 2019; 144(2): 482-493.e487.
- Freund Y, et al: FEBS Lett 2012; 586: 3410-3414.
- Dong C, et al: J Pharmacol Exp Ther 2016; 358: 413-422.
- Jarnagin K, et al: J Drugs Dermatol 2016; 15(4): 390-396.
- Zane LT, et al: Pediatr Dermatol 2016: 33(4): 380-387.
DERMATOLOGIE PRAXIS 2023; 30(1): 5–11









