
Un diagnóstico confirmado, pacientes convencidos del sentido y la finalidad del tratamiento, esto puede sonar banal, pero con respecto a un tratamiento farmacológico posiblemente de por vida, estos aspectos son muy relevantes. Además de una visión general compacta de las diferentes sustancias activas, se discuten las características especiales del tratamiento farmacológico en niños.
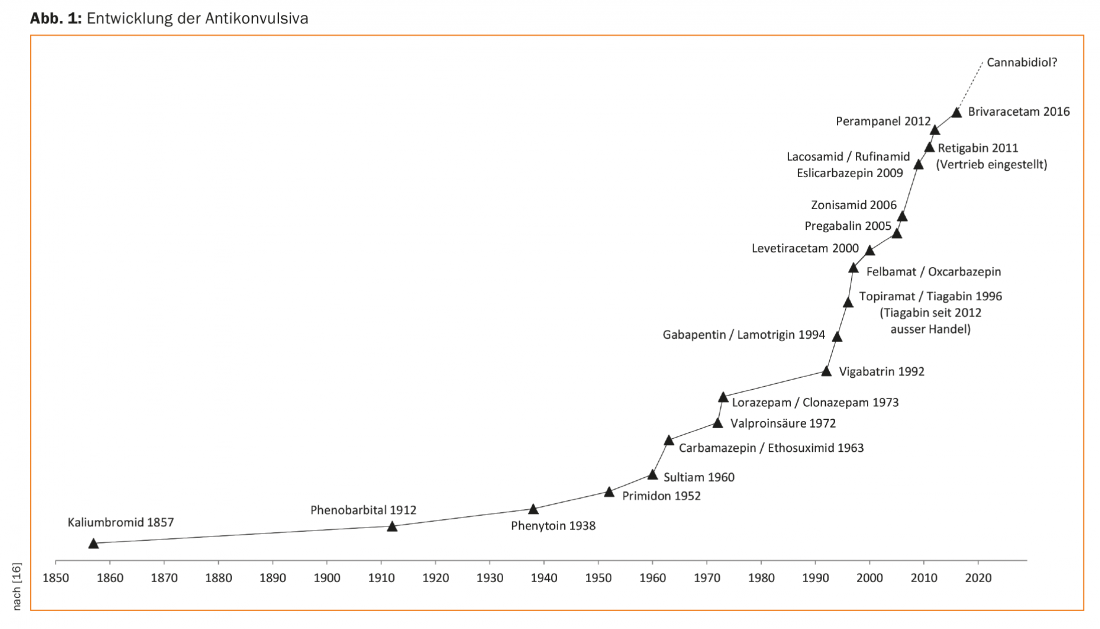
Las crisis epilépticas y las epilepsias son situaciones clínicas comunes y se conocen desde hace miles de años. Ya figuraba en tablillas cuneiformes de la época de los babilonios en el siglo II a.C. BC se registran los primeros textos médicos sobre epilepsias. A lo largo de los siglos, por tanto, se han utilizado un número y una variedad correspondientemente grandes de procedimientos para tratar las crisis epilépticas. Aunque Hipócrates ya había escrito hacia el 500 a.C. La epilepsia se consideraba una enfermedad originada en el cerebro, pero este conocimiento se olvidó y por ello durante mucho tiempo (y aún hoy en algunas regiones del mundo) los pacientes con epilepsia fueron considerados poseídos por el demonio y tratados con exorcismos y otras medidas justificadas religiosamente. Sólo en los siglos XVII y XVIII y sobre todo en el siglo XIX , poco a poco se impuso la visión científica actual [1]. El ginecólogo de la reina Victoria, Sir Charles Locock, describió por primera vez el efecto antiepiléptico del bromuro potásico en Lancet en 1857, estableciendo así el tratamiento farmacológico moderno de las epilepsias [2]. En 1912 se añadió otro fármaco, el fenobarbital, que todavía hoy desempeña un papel en el tratamiento de las epilepsias. Desde entonces, se han descubierto o probado varias sustancias. (Fig. 1), aunque todavía no se ha demostrado que ninguna de estas sustancias tenga un efecto antiepiléptico real, sino sólo un efecto anticonvulsivo, es decir, de supresión de las convulsiones. Por lo tanto, sería más correcto hablar de anticonvulsivos y no de antiepilépticos.
Para poder llevar a cabo una buena terapia farmacológica de la epilepsia, primero hay que confirmar el diagnóstico de la epilepsia. Esto puede sonar banal, pero no lo es en absoluto en la práctica clínica diaria, ya que a menudo, sobre todo al principio de la epilepsia, sólo se dispone de descripciones rudimentarias de las crisis y la tasa de diagnósticos erróneos es asombrosamente alta. En particular, el síncope causado, por ejemplo, por una arritmia cardiaca o una desregulación circulatoria también puede ir acompañado de descargas clónicas y simular así un ataque epiléptico tónico-clónico. Los trastornos funcionales o los trastornos hipercinéticos del movimiento, como la mioclonía, también deben tenerse en cuenta en el diagnóstico diferencial. Dado que el tratamiento de la epilepsia, especialmente en adultos, suele conllevar una terapia farmacológica de por vida y requiere un alto nivel de cumplimiento terapéutico, el paciente debe estar convencido del sentido y la finalidad del tratamiento. En caso de duda, debe considerarse la posibilidad de realizar un examen EEG por vídeo de larga duración para confirmar el diagnóstico.
Causas
Además de la historia clínica y la descripción de las crisis, para diagnosticar las epilepsias se utilizan principalmente el EEG y el diagnóstico por imagen, preferiblemente una RMNc. El progreso técnico de los dispositivos de RM también debe tenerse en cuenta aquí, de modo que si la RM es negativa, puede ser útil repetirla con un dispositivo más nuevo y realizarla con protocolos especiales de RM para epilepsia. El objetivo del diagnóstico es aclarar la causa o causas de la enfermedad con la mayor precisión posible. Se puede lograr la clasificación en un síndrome epiléptico, lo que influye en la selección del anticonvulsivo adecuado.
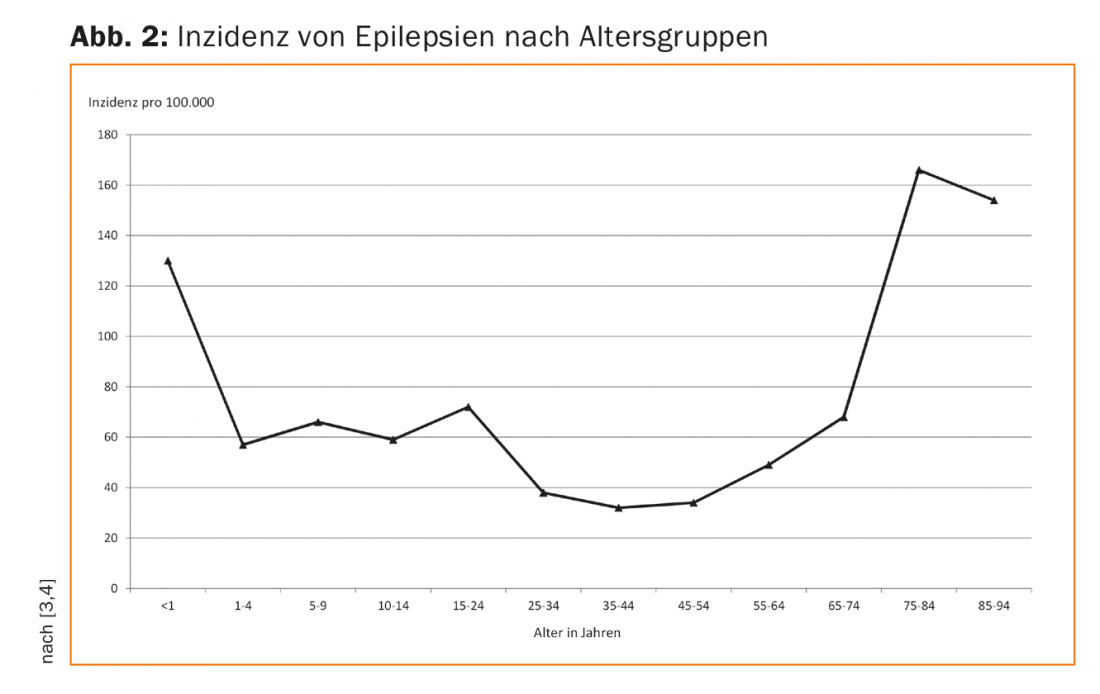
Naturalmente, las causas de la epilepsia en niños y adultos difieren en cierta medida. En los niños, por ejemplo, predominan las epilepsias debidas a una predisposición genética (por ejemplo, la epilepsia de ausencia y la epilepsia de Rolando), a malformaciones congénitas del cerebro o a la asfixia perinatal, y las convulsiones debidas a trastornos metabólicos (por ejemplo, las epilepsias dependientes de la vitamina B6). Las epilepsias debidas a daños cerebrales adquiridos también se dan en niños. Sin embargo, a diferencia de los niños, éstas representan con diferencia la mayor proporción de nuevas epilepsias en adultos. Debido al cambio demográfico, la incidencia de epilepsias en la vejez está aumentando significativamente, lo que conlleva nuevos retos en la terapia farmacológica en este grupo de edad (Fig. 2).
Epileptología pediátrica
En el contexto del diagnóstico y la terapia de las epilepsias infantiles, la etapa de desarrollo y la edad de los niños se añaden como dimensiones adicionales. Así pues, la frecuencia y el tipo de epilepsias difieren significativamente en los respectivos grupos de edad pediátrica. Históricamente, durante mucho tiempo no estuvo claro cómo clasificar los numerosos síndromes dentro del gran grupo de los trastornos convulsivos; en este sentido, el desarrollo del diagnóstico EEG por Hans Berger a mediados del siglo pasado supuso un gran avance. Algunos ejemplos son la epilepsia de Watanabe, el síndrome de West, la epilepsia de Rolando y la epilepsia de ausencias. La epilepsia de Watanabe, por ejemplo, son las convulsiones benignas del recién nacido que se producen en los tres primeros meses y desaparecen al final del primer año de vida. El síndrome de West fue descrito por primera vez por William James West en 1841 en Tonbridge, describiendo las características convulsiones flash Nick Salam. El síndrome de West es una epilepsia generalizada asociada a espasmos y suele aparecer entre los tres y los seis meses de edad. Se desencadena por una variedad de causas posibles (poliaetiología). El síndrome de West es un buen ejemplo de cómo diferentes causas conducen a una vía final fisiopatológica común y presentan así el mismo cuadro clínico electroencefalográfico y semiológico. Otro trastorno convulsivo pediátrico clásico es la epilepsia de Rolando, epilepsia focal idiopática benigna. También tiene una edad típica y se da sobre todo en niños pequeños y escolares. Es característico el inicio con hormigueo parestésico y expresiones motoras en la zona de la cara y no pocas veces con detención del habla. El EEG muestra las clásicas “ondas agudas” trifásicas sobre la región central. En los niños algo mayores se observa la clásica absencenepilepsia juvenil, que se acompaña de rigidez y falta de capacidad de respuesta a los estímulos. A menudo el diagnóstico de sospecha lo hacen los profesores porque los niños parecen ausentes en clase debido a que “sueñan despiertos”. El EEG muestra paroxismos generalizados de 3/s ondas espiga durante las ausencias.
De este extracto de síndromes epilépticos pediátricos ya se puede adivinar que la terapia debe ser específica según la caracterización del síndrome epiléptico [5].
Terapia farmacológica para niños
La elección del anticonvulsivo para la terapia a largo plazo en niños está decisivamente influida por la caracterización del trastorno convulsivo. Aparte del síndrome epiléptico, otros aspectos como el desarrollo y el objetivo de la terapia también son, por supuesto, decisivos para la elección. Sin embargo, para todas las epilepsias, el tratamiento agudo de un ataque depende de una benzodiacepina, normalmente midazolam o clonazepam. Existen formas orales, rectales y nasales de midazolam para el ámbito ambulatorio.
Para las epilepsias de Watanabe y las epilepsias infantiles benignas, se han popularizado la carbamazepina o la oxcarbazepina. El síndrome de West se trata clásicamente con un régimen que incluye vigabatrina y esteroides. La epilepsia de Rolando se trata clásicamente sobre todo con sultiam u oxcarbazepina, mientras que las ausencias suelen tratarse con etosuximida.
Además de estos síndromes epilépticos clásicos, también existen, por supuesto, epilepsias infantiles que son el resultado de cambios estructurales, por ejemplo, tras un accidente o una infección. Aquí se han establecido la lamotrigina, el levetiracetam o la oxcarbazepina, similares a los utilizados en adultos. En muchos otros síndromes epilépticos, como la epilepsia mioclónica juvenil o el síndrome de Lennox-Gastaut, así como en la epilepsia mioclónica-astásica, el valproato se ha establecido como el fármaco de primera elección [7].
La norma general en epileptología pediátrica es optar por la monoterapia si es posible para evitar efectos negativos en el desarrollo del niño [6]. Al considerar los objetivos del tratamiento, es importante tener en cuenta que el grado de movilidad del niño influye decisivamente en el objetivo del tratamiento. Por ejemplo, un niño que se mueve entre el tráfico o va a la piscina tiene un riesgo significativamente mayor de sufrir convulsiones que un niño inmóvil. Con el aumento de la edad y la independencia de los padres, también pasan a primer plano otros aspectos como la toma independiente de la medicación y el cumplimiento de la terapia.
En muchos síndromes epilépticos, un número no desdeñable de pacientes no sufrirá crisis ni siquiera sin medicación una vez completada la maduración cerebral, por lo que el epileptólogo pediátrico debe planificar un intento de cesación. Por lo tanto, debería discutirse con la familia en una fase temprana cuándo y cómo podría tener lugar un intento de salida, incluso si todavía quedan varios años en el futuro. Además, en el caso de los pacientes que dependerán de los anticonvulsivos durante el resto de su vida, es importante garantizar una atención continuada durante la transición de la pediatría a la medicina de adultos.
Terapia farmacológica para adultos
Una vez confirmado el diagnóstico de epilepsia, debe discutirse con el paciente qué síntoma diana debe tratarse. En la mayoría de los casos, se tratará de las “grandes” crisis tónico-clónicas de propagación bilateral (“gran mal”), que representan una carga considerable para los afectados, pero por supuesto también para sus familiares, y conllevan un riesgo de lesiones que puede llegar hasta la muerte súbita inesperada (SUDEP – “muerte súbita inesperada en pacientes con epilepsia”). Incluso las convulsiones puramente focales hasta entonces pueden dar lugar ocasionalmente a una gran convulsión tónico-clónica que se extiende bilateralmente con las correspondientes consecuencias. Algunos pacientes notan o no son conscientes de la no recuerdan sus convulsiones, por lo que el término “libre de convulsiones” suele ser difícil en la práctica clínica diaria. Crisis epilépticas repetidas resp. Una frecuencia elevada de convulsiones (incluidas las convulsiones subclínicas) también puede provocar trastornos cognitivos en el curso de la enfermedad [8]. Por lo tanto, la indicación del tratamiento debe discutirse con el paciente en cada caso concreto.
El tratamiento comienza entonces con un anticonvulsivo adecuado (véase más adelante), que se dosifica hasta alcanzar una dosis objetivo inicial. Si se producen nuevas convulsiones con esta dosis, deberá agotarse primero la dosis de este preparado hasta el umbral de tolerancia antes de establecer un cambio a otra sustancia o a una terapia combinada (add-on). El umbral de tolerancia varía mucho en función de la sustancia y del individuo.
En la actualidad se dispone de varias sustancias para el tratamiento de las epilepsias en adultos. Los agentes más utilizados en la actualidad son la lamotrigina, el levetiracetam, el valproato y la carbamazepina, y cada vez salen al mercado nuevos preparados. Sin embargo, las sustancias más nuevas no son necesariamente más eficaces en comparación con las antiguas, pero tienen muchos menos efectos secundarios y normalmente también menos interacciones con otros fármacos [7]. No obstante, en los grandes ensayos de fármacos, que suelen realizarse en pacientes con epilepsia refractaria a la terapia, una cierta proporción de pacientes (aprox. 10-20%) llegan a quedar libres de crisis con una nueva sustancia, por lo que bien puede estar justificado el correspondiente ensayo de terapia con una nueva sustancia.
Lamotrigina: Este preparado está disponible desde mediados de los años 90 y suele tolerarse muy bien sin efectos secundarios. Puede utilizarse tanto para las epilepsias focales (estructurales) como para las generalizadas. Se trata únicamente de un bloqueante oral de los canales de sodio. El principal inconveniente es que debe dosificarse muy lentamente para evitar las temidas reacciones alérgicas cutáneas, que pueden llegar hasta el síndrome de Lyell o de Stevens-Johnson, potencialmente mortales. La primera dosis objetivo habitual es de 150 mg (en ancianos) y 200 mg (dosis estándar) y sólo se alcanza al cabo de 4-6 semanas. Debido a su larga semivida de unas 24-35 horas, la lamotrigina también puede administrarse sólo una vez al día. Si es necesario, la dosis puede aumentarse a menudo de forma significativa hasta 500-800 mg al día durante el curso del tratamiento debido a su buena tolerabilidad [9].
Su uso durante el embarazo es posible y sólo se asocia a un ligero aumento del riesgo de malformaciones [10]. En la vida cotidiana, cabe señalar algunas interacciones farmacológicas relevantes de la lamotrigina: por ejemplo, el etinilestradiol de los anticonceptivos hormonales con componentes de estrógeno y progestágeno (“píldora”) puede reducir prácticamente a la mitad el nivel plasmático de lamotrigina, lo que, por un lado, puede conducir a una protección insuficiente contra las convulsiones mientras se toma la píldora y, por otro, a un aumento significativo de los niveles con los correspondientes efectos secundarios durante el descanso de la píldora. [11]. Por tanto, hay que aconsejar a las mujeres en consecuencia, y recomendamos un preparado sólo de progestágeno o un DIU para la terapia con lamotrigina. En el caso de las terapias combinadas, debe tenerse en cuenta sobre todo la interacción con el valproato. El valproato inhibe significativamente la descomposición de la lamotrigina, por lo que debe elegirse un régimen de dosificación ascendente mucho más lento y una dosis total de lamotrigina más baja [9]. Por otra parte, esta combinación suele tener mucho éxito debido a su lenta metabolización y, por tanto, a que sólo se producen pequeñas fluctuaciones en los niveles.
Levetiracetam: Este preparado puede utilizarse para epilepsias focales y también generalizadas y está disponible tanto en comprimidos como en jarabe y para uso intravenoso, por lo que desempeña un papel importante en la terapia aguda hospitalaria. La primera dosis objetivo es de 750 mg (ancianos) a 1000 mg (dosis estándar). Puede aumentarse hasta aprox. 4000 mg y se administra dos veces al día (semivida aprox. 7 horas). El efecto está mediado a través de la proteína de la vesícula presináptica SV2. El levetiracetam apenas se une a las proteínas plasmáticas y prácticamente no tiene potencial de interacción. Sin embargo, pueden aparecer síntomas psiquiátricos como irritabilidad, inquietud interior, agresividad, ansiedad y depresión como efectos secundarios importantes. En la literatura, la tasa de estos efectos secundarios se cifra en un 10-15%; en la práctica clínica, esta tasa parece ser aún mayor, del 20-30%, aunque no siempre conduce a un cambio de terapia. Por lo tanto, no es favorable su uso en pacientes con problemas de comportamiento y/o, por ejemplo, lesiones cerebrales frontales. El levetiracetam tiene un buen efecto sobre la mioclonía. Junto con la lamotrigina, es el fármaco de primera elección en el embarazo.
Otro desarrollo del levetiracetam es el brivaracetam (Briviact®), que fue aprobado como el anticonvulsivo más nuevo en Suiza a finales de 2016. Este preparado tiene menos efectos secundarios psiquiátricos que el levetiracetam con (al menos) el mismo buen efecto, la dosis objetivo es de 100-200 mg, dividida en dos dosis, se puede dosificar rápidamente resp. la dosis objetivo puede iniciarse directamente. Cuando cambie del levetiracetam al brivaracetam (por ejemplo, debido a síntomas psiquiátricos), el cambio puede hacerse “de la noche a la mañana” en una proporción de 10-15 a 1 (por ejemplo, 1000 mg de levetiracetam por 100 mg de brivaracetam).
Valproato: El valproato es una sustancia que se descubrió en los años 60 y que sigue desempeñando un papel importante en el tratamiento de las epilepsias. Probablemente actúa potenciando los mecanismos GABAérgicos y se utiliza tanto en las epilepsias focales como en las generalizadas. Especialmente para esta última, a menudo se considera la terapia más eficaz. La primera dosis objetivo habitual es de 1000 mg, dividida en dos tomas, aunque el preparado de liberación sostenida también puede administrarse una sola vez al día. A diferencia de la mayoría de los demás anticonvulsivantes, el valproato se metaboliza predominantemente por vía hepática, por lo que también puede utilizarse en caso de insuficiencia renal. Los efectos secundarios más relevantes son un aumento de peso a menudo significativo y una posible ralentización cognitiva. El valproato es un potente inhibidor enzimático, por lo que deben tenerse en cuenta las interacciones relevantes durante el tratamiento en la práctica clínica. Cuando se combina con lamotrigina, se inhibe su degradación, lo que aumenta significativamente el nivel de lamotrigina y puede provocar signos de intoxicación (véase más arriba). La terapia antibiótica con carbapenem puede causar un descenso significativo de los niveles de valproato en un plazo de 24 a 48 horas, lo que puede provocar un estado epiléptico [12]. El metabolismo en el hígado puede conducir a un aumento de la producción de amoníaco, que puede causar encefalopatía. Para las mujeres en edad fértil, resp. No debe utilizarse del mismo modo debido a su considerable teratogenicidad. sólo debe utilizarse tras una evaluación detallada de los riesgos y beneficios y una declaración de consentimiento por escrito de la mujer afectada (véase también la sección Embarazo).
Carbamazepina: La carbamazepina también se descubrió en la década de 1960. Es un bloqueante de los canales de sodio utilizado para las epilepsias focales. En las epilepsias generalizadas, puede producirse un empeoramiento de la situación convulsiva, especialmente las mioclonías y las ausencias. La primera dosis objetivo habitual es de 800 mg, dividida en dos dosis (semivida por autoinducción de 16-24 horas, con una dosis única de unas 36 horas [9]). El efecto secundario más relevante de la carbamazepina es el mareo, a veces con nistagmo, debido a su estrecho margen terapéutico. Las reacciones cutáneas tampoco son infrecuentes con la carbamazepina, que se asocia a alelos HLA específicos, presentes sobre todo en japoneses, chinos y tailandeses, pero algo menos frecuentemente en caucásicos. Como la carbamazepina es un fuerte inductor enzimático y, por tanto, existen muchas interacciones farmacológicas, su uso en pacientes ancianos suele estar limitado, aunque en sí misma tiene un buen efecto anticonvulsivo. Puede utilizarse en el embarazo, aunque las tasas de aborto son algo más elevadas que con la lamotrigina y el levetiracetam [10].
Situaciones especiales – Embarazo
Existe una necesidad especial de asesoramiento para las mujeres jóvenes en edad fértil con epilepsias con respecto a un posible embarazo. La epilepsia per se no es una razón para no quedarse embarazada y en más del 90% de los casos las mujeres con epilepsia suelen dar a luz a niños sanos sin complicaciones. Sin embargo, hay algunas cosas importantes que debe tener en cuenta [13]. La sustitución con ácido fólico debe iniciarse ya si se desea tener un hijo, es decir, antes de que se produzca el embarazo (véase también el artículo sobre la epilepsia y el deseo de tener hijos en la página 18 de este número).
Lo más importante es, sin duda, el ajuste óptimo de la medicación para proteger a la mujer y al feto de las convulsiones durante el embarazo. La primera elección en mujeres embarazadas son la lamotrigina y el levetiracetam, que sólo mostraron un ligero aumento del riesgo de malformaciones en grandes estudios prospectivos de registro (por ejemplo, el registro EURAP) [10]. A diferencia de la lamotrigina y el levetiracetam, el riesgo de malformaciones es muy elevado con el valproato (dependiente de la dosis hasta aproximadamente el 45%(!), especialmente defectos del tubo neural, pero también alteraciones cognitivas del niño [14]). En este caso, el efecto teratogénico se produce sobre todo en las primeras semanas de embarazo, cuando la mujer a menudo ni siquiera sabe que está embarazada, por lo que el valproato no debe utilizarse en mujeres en edad fértil si es posible; las autoridades reguladoras han emitido las advertencias correspondientes (“letra roja”). Sería deseable incluir a todas las mujeres con epilepsias y una terapia con anticonvulsivos en un registro de embarazos como el registro EURAP antes mencionado para obtener afirmaciones aún más fiables sobre la teratogenicidad de cada una de las sustancias. Cualquier médico puede descargar de Internet los formularios correspondientes, rellenarlos y enviarlos a un centro de estudios adecuado.
Límites de la terapia – cirugía de la epilepsia
Aunque la ausencia de crisis suele ser el objetivo principal del tratamiento, sólo se consigue en un 60-70% de los pacientes por término medio, dependiendo del síndrome epiléptico. Incluso con terapias combinadas, sólo unos pocos pacientes quedan además libres de convulsiones. Estas tasas no han aumentado significativamente en las últimas décadas, ni siquiera con los nuevos fármacos. Por lo tanto, además de comprobar el diagnóstico de epilepsia y su cumplimiento, también debe considerarse la cirugía de la epilepsia en una fase temprana. Esto puede conducir a la recuperación permanente de la epilepsia en pacientes adecuados con epilepsias focales hasta en un 80-90%, dependiendo del origen de las crisis. En los últimos años, por tanto, se ha iniciado aquí un replanteamiento que va desde la cirugía de la epilepsia como último recurso hacia una terapia de (casi) primera elección (por ejemplo, en el caso de la epilepsia del lóbulo temporal mesial con esclerosis del hipocampo – véase también el artículo el artículo sobre el tratamiento quirúrgico en la página 26 de este número. [15]).
Mientras que en algunas epilepsias infantiles y adolescentes se sabe que la enfermedad “se cura”, esto rara vez ocurre en la edad adulta. No obstante, un intento prudente de interrupción puede estar justificado en determinadas circunstancias (¿diagnóstico erróneo de DD en el pasado?). Por otro lado, si la causa de la epilepsia persiste (por ejemplo, malformación, defecto postraumático o postisquémico) y el paciente está libre de crisis, esto no debe ponerse en peligro.
Desde un punto de vista pragmático, reducimos la dosis a una dosis baja en las epilepsias generalizadas, a largo plazo libres de crisis y a petición explícita del paciente, mientras realizamos controles EEG (el EEG es más adecuado para predecir el riesgo de recurrencia de crisis en las epilepsias generalizadas que en las focales). A continuación, llevamos a cabo el último paso de la dosificación bajo una monitorización de vídeo-EEG a largo plazo, pero, por supuesto, sigue existiendo un cierto riesgo residual de que se produzca una convulsión de recaída, que el paciente debe estar dispuesto a soportar.
Mensajes para llevarse a casa
- Debe confirmarse el diagnóstico de epilepsia.
- La frecuencia y el tipo de epilepsias difieren significativamente en los grupos de edad pediátrica.
- La elección del fármaco antiepiléptico para la terapia a largo plazo en niños está decisivamente influida por la caracterización del trastorno convulsivo.
- En muchos síndromes epilépticos, los pacientes estarán libres de crisis sin medicación una vez que se haya completado la maduración cerebral, por lo que el intento de abandono también debe planificarse a largo plazo.
- En epileptología pediátrica, la monoterapia debe aplicarse siempre que sea posible.
- Las dosis deben aumentarse desde la primera dosis objetivo en función de las convulsiones hasta el límite de tolerancia; sólo entonces cambiar a otro preparado o complemento (terapia combinada/para adultos).
- Deben tenerse en cuenta las interacciones con otros medicamentos: La carbamazepina es un inductor enzimático, el valproato es un inhibidor enzimático, los carbapenems pueden reducir drásticamente los niveles de valproato.
- Si es posible, el valproato no debe utilizarse en mujeres en edad fértil.
Literatura:
- Gonzalo A (ed.): Introducción a la epilepsia. Cambridge University Press; 2012.
- Brody MJ: Terapia con fármacos antiepilépticos: la historia hasta ahora. Convulsiones 2010 dic; 19(10): 650-655.
- Werhahn KJ: Epilepsia en ancianos. Dtsch Arztebl Int. 2009; 106(9): 135-142.
- Olafsson E, et al: Incidencia de crisis no provocadas y epilepsia en Islandia y evaluación de la clasificación del síndrome epiléptico: un estudio prospectivo. Lancet Neurol 2005; 4: 627-634
- Broser P, Maier O: Encefalopatías epilépticas infantiles precoces. Epileptología 2016; 33: 95-101.
- Pellock JM, Bourgeois BFD, Dodson WE (eds.): Epilepsia pediátrica. Nueva York, Demos, 2007.
- Steinhoff B, Bast T: Vademécum antiepiléptico. Sociedad Alemana de Epileptología 2017.
- Vingerhoets G: Efectos cognitivos de las convulsiones. Convulsiones 2006; 15(4): 221-226.
- Swissmedic: www.compendium.ch. Recuperado en julio de 2018
- Tomson T, et al: Riesgo comparativo de malformaciones congénitas mayores con ocho fármacos antiepilépticos diferentes: un estudio prospectivo de cohortes del registro EURAP. Lancet Neurol 2018 Jun; 17(6): 530-538.
- Sabers A, et al: Niveles plasmáticos de lamotrigina reducidos por los anticonceptivos orales. Epilepsy Res 2001 Nov; 47(1-2): 151-144.
- Sutter R, Rüegg S, Tschudin-Sutter S: Convulsiones como efectos adversos de los fármacos antibióticos: una revisión sistemática. Neurología 2015 Oct 13; 85(15): 1332-13341.
- Voinescu PE, Penell PB: Tratamiento de la epilepsia durante el embarazo. Expert Rev. Neurother 2015; 15(10): 1171-1187.
- Meador KJ et al: Exposición fetal a fármacos antiepilépticos y resultados cognitivos a la edad de 6 años (estudio NEAD): un estudio observacional prospectivo. Lancet Neurol 2013 Mar; 12(3): 244-252.
- Steinhoff BJ, Staack AM: ¿Hay lugar para el tratamiento quirúrgico de la epilepsia no farmacorresistente? Epilepsia Behav 2018.
- Golyala A, Kwan P: Desarrollo de fármacos para la epilepsia refractaria: Los últimos 25 años y más allá; Seizure 2017 Jan; 44: 147-156.
InFo NEUROLOGÍA Y PSIQUIATRÍA 2018; 16(5): 12-17.









