Las dermatosis ampollosas autoinmunes (BAID) son un grupo de enfermedades raras, potencialmente mortales, que se manifiestan clínicamente como lesiones en la piel o las mucosas, pero que, contrariamente a lo que se intuye, no siempre tienen por qué ir acompañadas de ampollas. Los cambios en la piel y las mucosas se desencadenan por una reacción del sistema inmunitario mediada por autoanticuerpos contra proteínas estructurales de la piel.
Las dermatosis ampollosas autoinmunes (BAID) son un grupo de enfermedades raras, potencialmente mortales, que se manifiestan clínicamente como lesiones en la piel o las mucosas, pero que, contrariamente a lo que se intuye, no siempre tienen por qué ir acompañadas de ampollas. Los cambios cutáneos y mucosos se desencadenan por una reacción del sistema inmunitario mediada por autoanticuerpos (AAK) contra proteínas estructurales de la piel que provocan la integridad del propio epitelio (desmosomas) o la del anclaje epitelial al tejido conectivo subyacente de la piel o la mucosa. Las primeras se agrupan como enfermedades del pénfigo y las segundas como enfermedades del penfigoide (Tab. 1) [1,2]. Debe distinguirse de la dermatitis herpetiforme, en la que se forman anticuerpos contra las transglutaminasas 2 y 3 y que siempre va acompañada de enfermedad celíaca [3].
El antígeno diana de los autoanticuerpos influye decisivamente en la patogénesis y, por tanto, en el aspecto clínico de la BAID, sirve mediante su determinación para establecer un diagnóstico exacto e influye decisivamente en el pronóstico y la terapia a aplicar. A continuación, se considerarán en particular el reconocimiento y la diferenciación de las enfermedades penfigus y penfigoide, así como los pasos del diagnóstico. Encontrará información más detallada en las directrices actuales [4,5] (directriz de la EADV sobre el pénfigo y la DH), así como en los artículos de investigación científica y las revisiones que se publican periódicamente.
Epidemiología: A excepción del penfigoide gestationis, todas las BAID pueden aparecer en principio a cualquier edad, pero existe una distribución por edades típica de cada una de las enfermedades. Mientras que la dermatosis IgA lineal es más común en niños y adolescentes, las personas con penfigoide bulloso suelen desarrollar la enfermedad después de los 75 años, y los pacientes con pénfigo son por término medio una o dos décadas más jóvenes. [6,7]. Geográficamente, el penfigoide bulloso predomina en el norte y centro de Europa y Norteamérica, con una incidencia de unos 20/millón de habitantes/año. En el sur de Europa, Israel e Irán, el pénfigo es la BAID más común, con una incidencia anual de 5-15 millones/habitante [1]. En Suiza, la incidencia del penfigoide bulloso fue de 12,1/millón/año y la del pénfigo vulgar y foliáceo de 0,6/millón/año en 2001/2002 [8]. Todas las demás BAID se dan incluso con menos frecuencia que el pénfigo.
Autoantígenos y presentación clínica
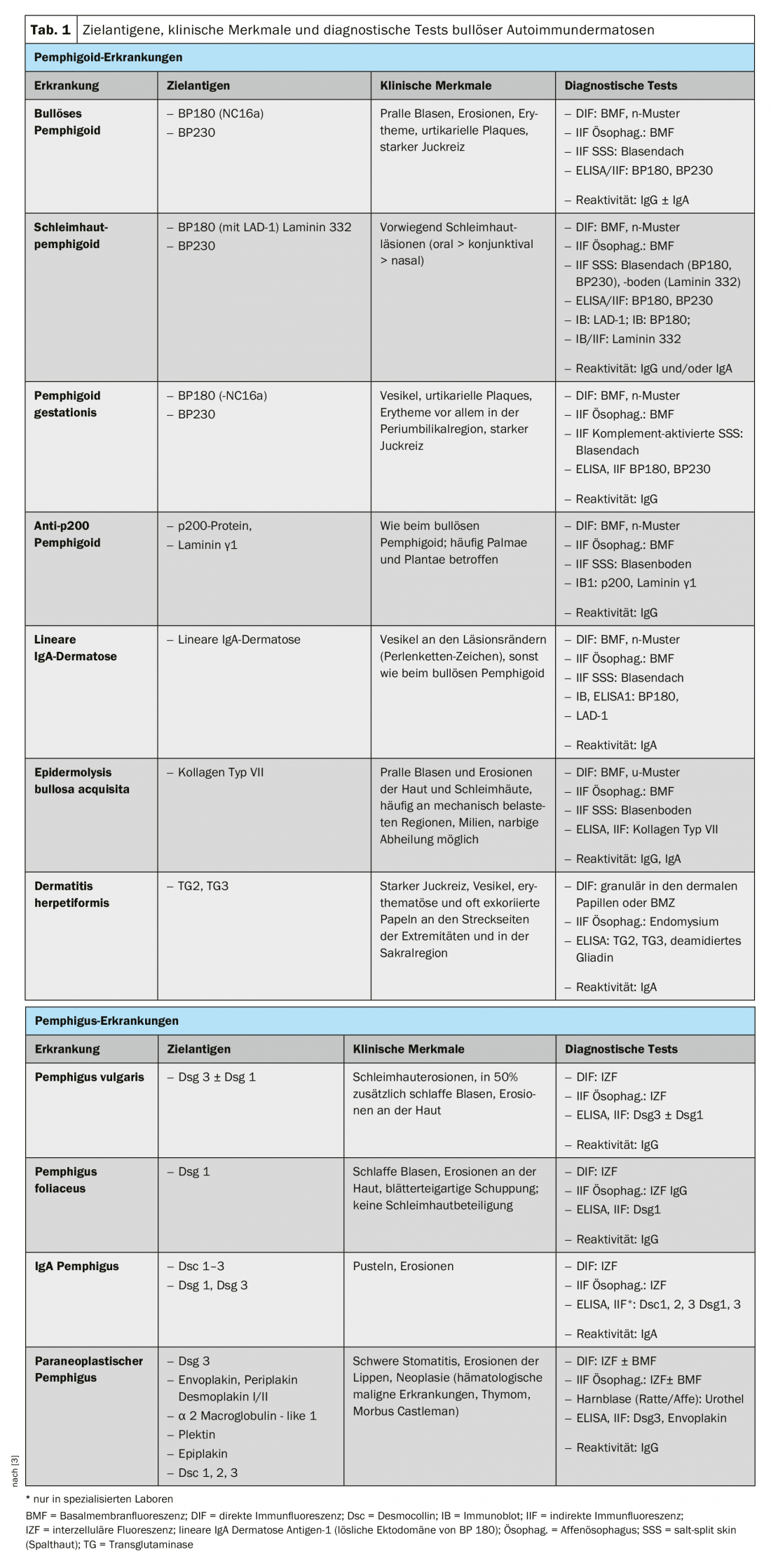
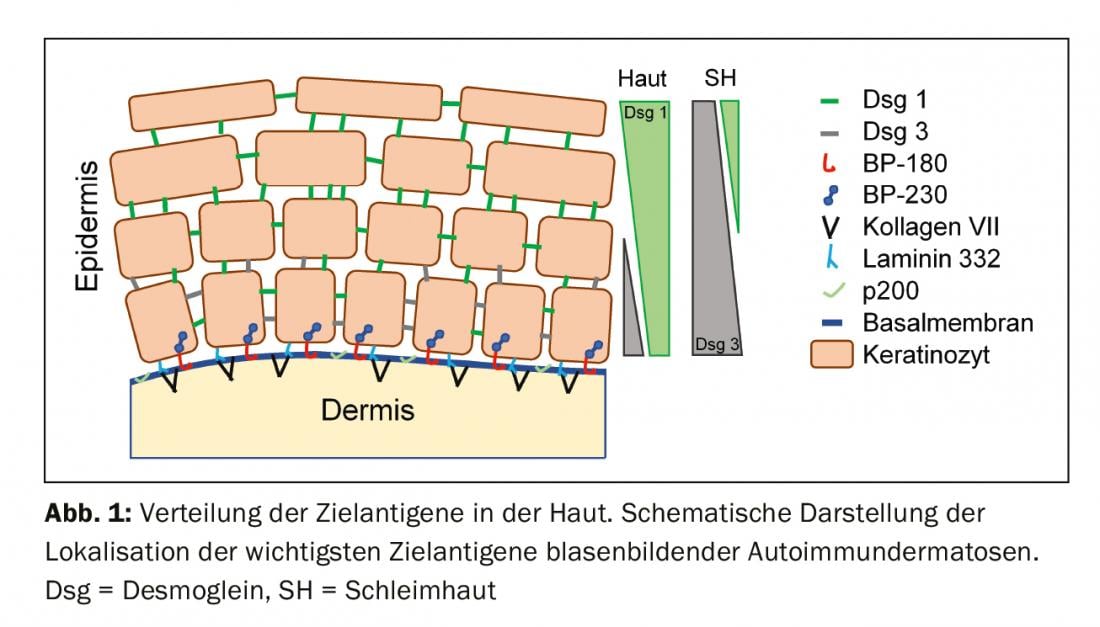
Para comprender el cuadro clínico de la BAID, conviene fijarse primero en la distribución de los antígenos diana dentro de la piel y las mucosas (Fig. 1) . Los antígenos diana del grupo penfigoide aseguran el anclaje del epitelio a la membrana basal y al tejido conjuntivo subyacente. Esta zona también se denomina zona de unión dermoepidérmica. Cuando los autoanticuerpos se unen a estas proteínas estructurales, se produce una compleja reacción inflamatoria con activación local del sistema del complemento, infiltración de eosinófilos, neutrófilos, macrófagos y linfocitos en la dermis superior y posterior liberación de especies reactivas de oxígeno y proteasas por parte de estas células, que acaban destruyendo la zona de unión dermoepidérmica [9]. Esto da lugar a la formación de hendiduras subepidérmicas y ampollas abultadas (serosas/hemorrágicas) clínicamente típicas (Fig. 2C). La irritación mecánica provoca la erosión de la piel y, a veces, de las mucosas. Los autoanticuerpos del penfigoide ampolloso se unen al BP180 (colágeno tipo XVII), y en particular al dominio NC16A del BP180, y en la mitad de los pacientes también al BP230 (tabla 1) . El 20% de los casos de penfigoide ampolloso evolucionan sin formación de ampollas, con una variedad de cuadros clínicos, como eritema urticarial, pápulas excoriadas y eccema [10]. Prácticamente siempre hay un picor pronunciado. De gran importancia clínica es la asociación de la PA con diversas enfermedades neurológicas en un 30-50% de los pacientes, así como con la diabetes mellitus y las neoplasias hematológicas [2,11]. Recientemente se ha puesto de manifiesto que ciertos fármacos antidiabéticos orales, los inhibidores de la dipeptidil peptidasa IV, en particular la vildagliptina, pueden desencadenar la PA, por lo que estos fármacos no deben utilizarse en la PA [12].
El dominio NC16A de la BP180 es también la región inmunodominante en el penfigoide gestacional, una BAID que se produce predominantemente en la segunda mitad del embarazo y suele resolverse 5-6 meses después del parto. Sin embargo, las recidivas son frecuentes en los nuevos embarazos. La dermatosis IgA lineal presenta anticuerpos IgA contra más epítopos C-terminales del BP180.
En el penfigoide mucoso, se ven afectadas principalmente las membranas mucosas, incluidas la cavidad oral y la conjuntiva, así como las membranas mucosas de la nariz, la garganta, la faringe , la tráquea, el esófago y los genitales (Fig. 2D) . Excepto en la boca, se producen cicatrices con regularidad. Las complicaciones de esta cicatrización incluyen la posible ceguera y la obstrucción de las vías respiratorias. En el penfigoide mucoso, los autoanticuerpos se dirigen contra epítopos C-terminales de BP180 o laminina 332 (Tab. 1) . El penfigoide anti-p200 es clínicamente similar al PA, pero los autoanticuerpos se dirigen contra una proteína de 200 kDa de la unión dermoepidérmica. En la gran mayoría de los pacientes, los autoanticuerpos también reaccionan con la laminina γ1 [13]. La epidermólisis ampollosa adquirida se define por la presencia de anticuerpos contra el colágeno de tipo VII y se manifiesta clínicamente de dos formas principales, la variante inflamatoria parecida a un PA o penfigoide mucoso, o la variante mecanoampollosa clásica con ampollas, erosiones y cicatrices en zonas corporales expuestas como el dorso de la mano [14].
Los antígenos diana del grupo del pénfigo, las cadherinas desmosomales desmogleína 1 y 3 (Dsg 1, 3), son formados por las células epiteliales para unirse firmemente entre sí (Fig. 1). Si son atacadas, la asociación de células epiteliales se disuelve (acantólisis). Clínicamente, esto puede demostrarse utilizando fuerzas de cizallamiento para desprender superficialmente la piel perilesional de aspecto sano (fenómeno de Nikolski). Sin embargo, dado que la estabilidad de una ampolla depende del grado de integridad intraepitelial y ésta está directamente dañada, en el pénfigo se observan ampollas flácidas que se rompen rápidamente, por lo que suelen formarse erosiones. El Dsg 1, el antígeno diana del pénfigo foliáceo (PF), se produce principalmente en las capas epidérmicas superiores de la piel y las mucosas, aunque el Dsg 3 no se expresa allí en la piel, por lo que el PF se manifiesta por erosiones, costras y una fina descamación laminar en la piel, pero las mucosas están siempre libres (hipótesis de compensación del Dsg, Fig. 2A) [1,15]. El Dsg 3, el antígeno diana en el pénfigo vulgar (PV), se expresa principalmente en las capas inferiores del epitelio; sin embargo, en estos lugares el Dsg1 sólo se encuentra en la mucosa epitelial pero no en la piel. Así, en presencia exclusiva de anticuerpos anti-Dsg3, la PV desarrolla exclusivamente erosiones mucosas (Fig. 2B), que suelen ser dolorosas, impiden la ingesta de alimentos y pueden provocar pérdida de peso. En la PV mucocutánea con anticuerpos contra Dsg3 y Dsg1, se producen erosiones y ampollas flácidas en la piel además de las lesiones mucosas. A diferencia de las enfermedades penfigoides, no es necesaria ninguna reacción inflamatoria para la formación de ampollas; la unión de los anticuerpos anti-Dsg a las células epiteliales basta por sí sola para desencadenar la acantólisis. El impedimento estérico de la interacción desmosómica por la unión de los autoanticuerpos, una expresión alterada de las moléculas Dsg en la superficie celular así como una compleja remodelación del citoesqueleto de las células epiteliales mediada por la transducción de señales desempeñan aquí un papel decisivo [15]. El pénfigo paraneoplásico se asocia siempre a una neoplasia, normalmente un timoma o una neoplasia hematológica, y se caracteriza clínicamente por una estomatitis marcada y una afectación frecuente de los labios. Además del Dsg 3, los autoanticuerpos se dirigen contra proteínas de la familia de la plakina (incluyendo la envoplakina, la periplakina, la BP230) y contra la α2-macroglobulina-like 1 [1].
Diagnóstico
Dado que la presentación clínica de la BAID es muy variada y no siempre se puede atribuir claramente a una BAID específica, el diagnóstico diferencial debe excluir las causas infecciosas (impétigo, erisipela bullosa, síndrome de la piel escaldada estafilocócica, infecciones por virus herpes), las enfermedades hereditarias (porfiria, epidermólisis bullosa hereditaria), así como el exantema por fármacos y las noxas químicas y físicas [4]. En caso de afectación de la cavidad oral, el diagnóstico diferencial debe ser principalmente un liquen ruber mucosoe, y en caso de afectación ocular, debe considerarse una génesis infecciosa, alérgica o irritativa. Especialmente en caso de afectación ocular, debe considerarse precozmente la posibilidad de un penfigoide mucoso, ya que es la única forma de prevenir terapéuticamente la cicatrización irreversible. La figura 2 y la tabla 1 resumen las características clínicas.
Además de los hallazgos clínicos, la inmunofluorescencia (IF) directa de una biopsia perilesional y la detección de autoanticuerpos circulantes mediante IF indirecta, ensayo inmunoenzimático (ELISA) e inmunoblots son esenciales en el diagnóstico de la BAID [4].
El diagnóstico exacto de cada BAID tiene importancia pronóstica y es relevante para la terapia. Así pues, el penfigo paraneoplásico se asocia prácticamente siempre a malignidad y el penfigoide mucinoso anti-laminina-332 se asocia a malignidad en el 25-30% de los casos y en estos pacientes está indicada la búsqueda de tumores [1,16,17]. Las enfermedades del penfigo, la epidermólisis bullosa adquirida y el penfigoide mucoso con afectación ocular, laríngea o traqueal sólo pueden tratarse con una terapia inmunosupresora intensiva, mientras que el penfigoide anti-p200, las dermatosis IgA lineales y la dermatitis herpetiforme suelen requerir sólo una inmunosupresión leve o una dieta sin gluten.
Inmunofluorescencia directa
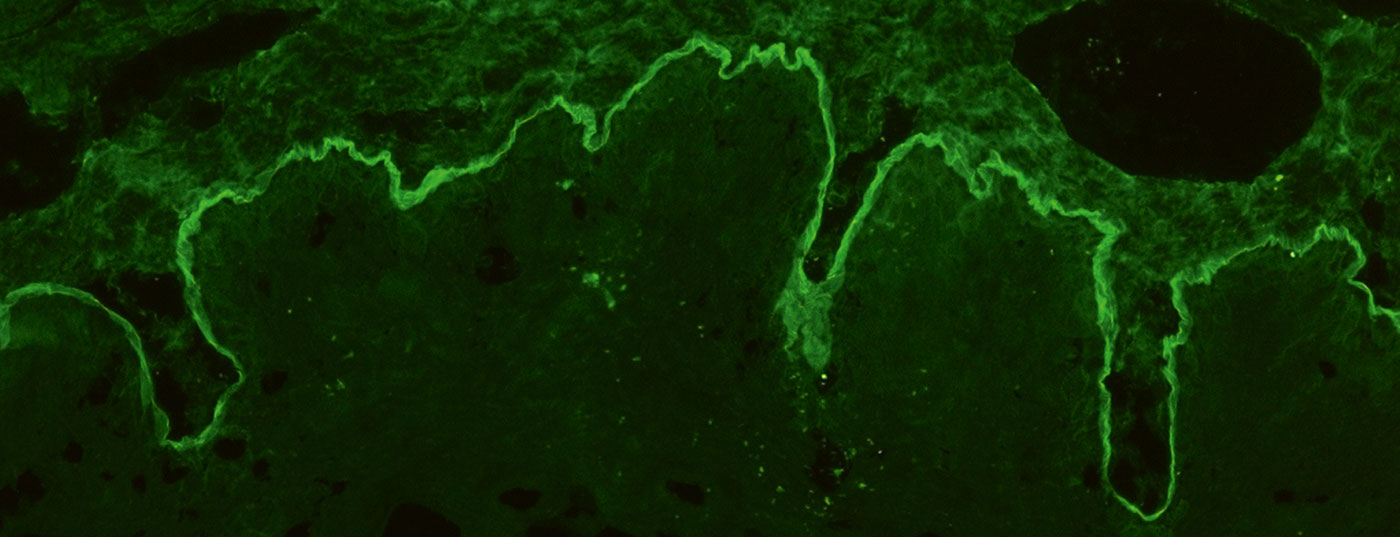
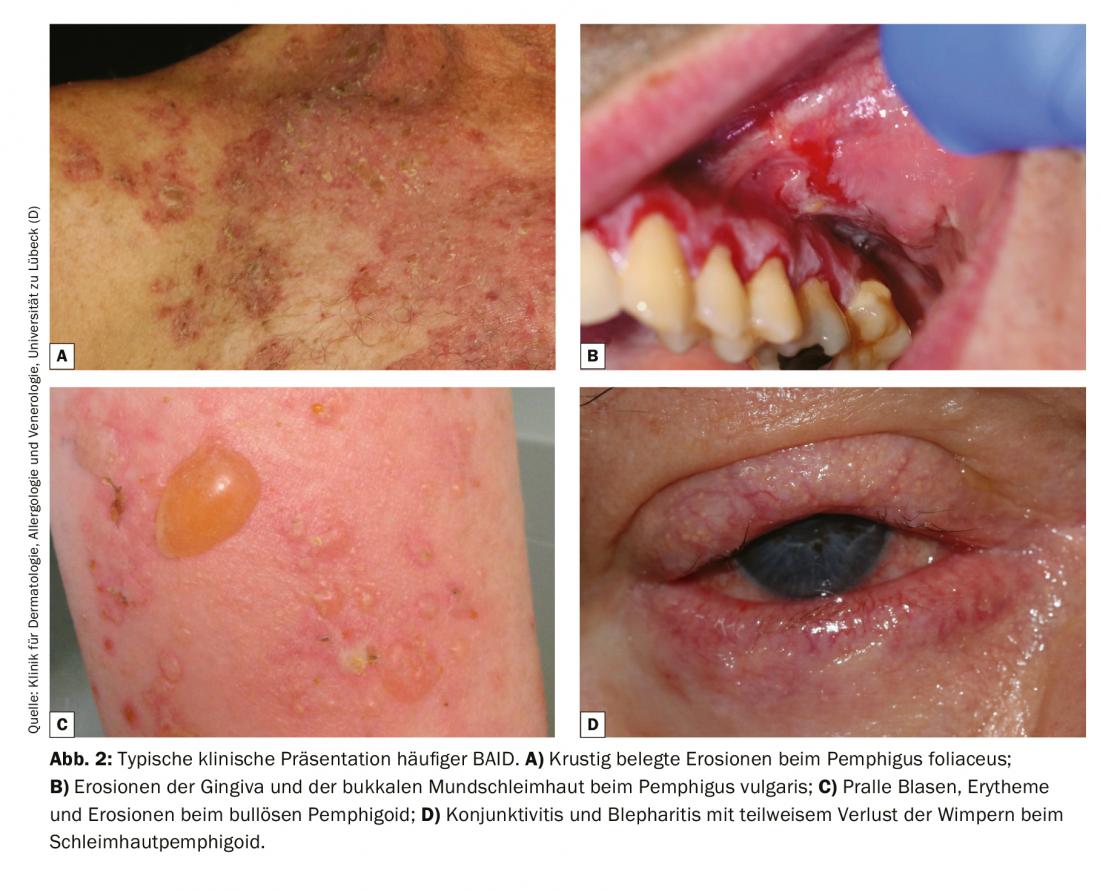
La IF directa de una biopsia perilesional sigue siendo el patrón oro diagnóstico de la BAID con una especificidad del 98% y una sensibilidad del 80-90% [4]. La biopsia de la muestra debe congelarse inmediatamente después de la recogida o almacenarse en NaCl al 0,9% o en medio Michels y procesarse en un plazo de 72 horas. A continuación, los autoanticuerpos unidos al tejido (inmunoglobulina [Ig] G, A y M) y el factor del complemento C3 se visualizan en secciones congeladas con un anticuerpo marcado con fluorescencia. El reconocimiento de un patrón de fluorescencia típico permite básicamente el diagnóstico fiable de pénfigo (patrón de fluorescencia/red intercelular epitelial), penfigoide (fluorescencia lineal a lo largo de la membrana basal) y dermatitis herpetiforme (depósitos granulares de IgA en las puntas papilares o a lo largo de la membrana basal, Fig. 3). Además, debe tomarse una biopsia lesional para realizar un examen histológico; esto sirve para diagnosticar otras enfermedades, especialmente en caso de IF directa y serología negativas.
Inmunofluorescencia indirecta en sustratos de órganos
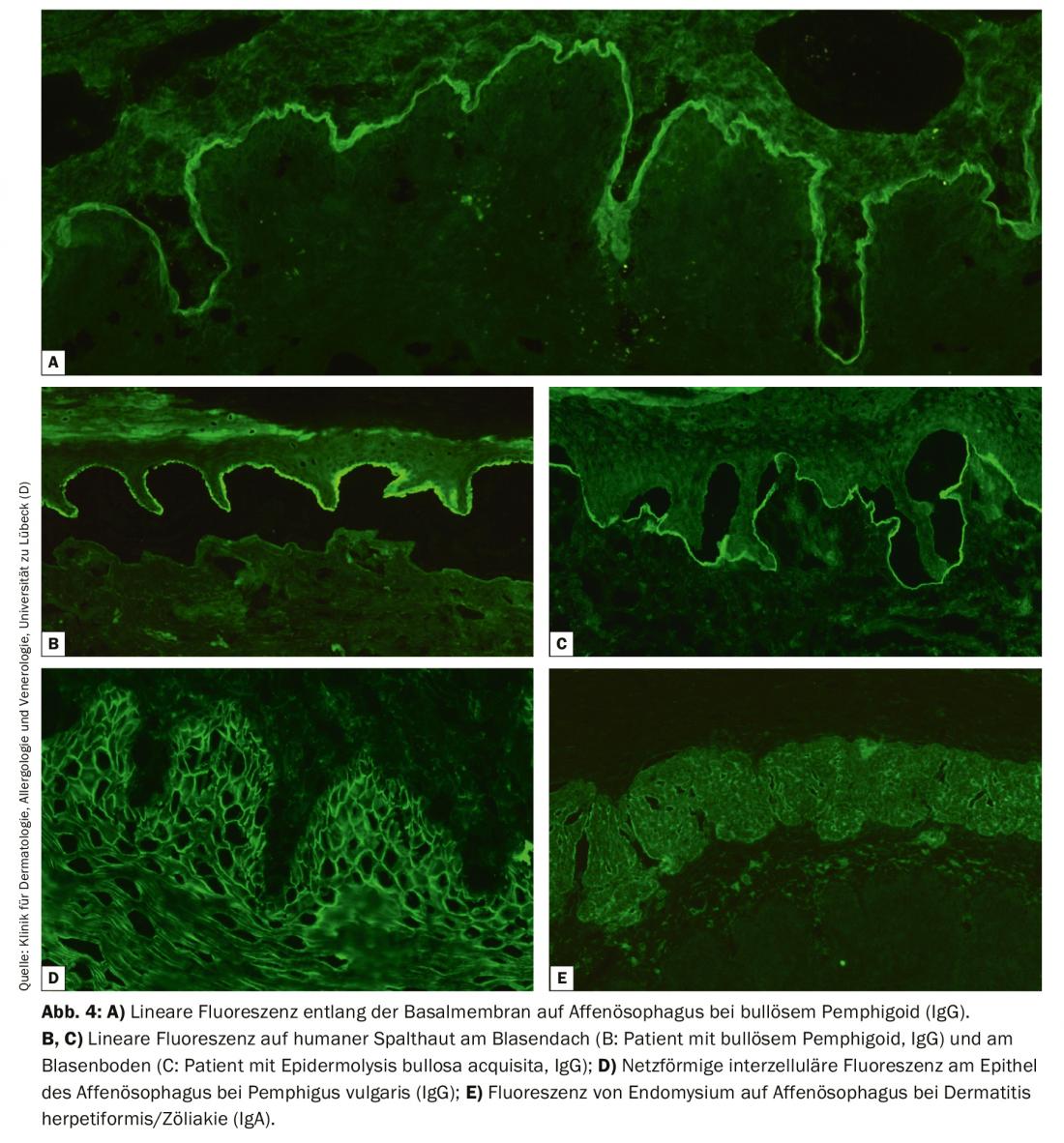
Para la IF indirecta se utiliza sobre todo el esófago de primates y con solución de NaCl. La piel humana dividida en la membrana basal, denominada piel dividida, se utiliza para la detección de autoanticuerpos séricos en la BAID. En la sección esofágica, pueden detectarse autoanticuerpos a lo largo de la membrana basal en las enfermedades penfigoides. (Fig. 4A), en las enfermedades del pénfigo intercelularmente en el epitelio (Fig. 4D) y en la dermatitis herpetiforme y la enfermedad celíaca contra el endomisio (sólo IgA, Fig. 4E). En la piel dividida, los autoanticuerpos séricos de las enfermedades penfigoides se unen bien en el techo de la ampolla creada artificialmente (en BP, penfigoide mucoso anti-BP180, penfigoide gestationis, dermatosis IgA lineal, Fig. 4B) o en el suelo de la ampolla (en el penfigoide anti-p200, el penfigoide mucoso anti-laminina 332, la epidermólisis bullosa adquirida, Fig. 4C). Aquí también buscamos depósitos de IgG e IgA, siendo estos últimos típicos de la dermatosis IgA lineal, por ejemplo. Con menor frecuencia se indica el examen de la vejiga de mono/rata en el pénfigo paraneoplásico o la prueba de fijación del complemento en piel escindida en la sospecha de penfigoide gestationis.
La IF indirecta en los sustratos orgánicos mencionados se utiliza como prueba de cribado y, a continuación, en función del resultado, se realizan pruebas antígeno-específicas, ELISA, IF indirecta, inmunoblots o inmunoprecipitación, si es necesario, teniendo en cuenta la IF directa. Con el desarrollo de pruebas antígeno-específicas, que utilizan sobre todo las formas inmunodominantes recombinantes de los autoantígenos, el diagnóstico puede hacerse ahora serológicamente en el 80-90% de los pacientes con BAID [18–20].
ELISA
Existen ELISA comerciales muy sensibles y específicos para los principales antígenos diana, Dsg1, Dsg3, Envoplakin, BP180 NC16A, BP230 y colágeno tipo VII [3]. En el caso de los anticuerpos contra Dsg1, BP180 NC16A, colágeno tipo VII y, en menor medida, Dsg3, se ha demostrado que los niveles séricos se correlacionan bien con la actividad clínica en la piel y las mucosas de pacientes con pénfigo, penfigoide ampolloso y epidermólisis ampollosa adquirida, de modo que los ELISA también pueden utilizarse para controlar la actividad de la enfermedad durante su curso. Esto es especialmente relevante para los pacientes en remisión clínica, en los que debe decidirse hasta qué punto puede reducirse aún más la terapia inmunosupresora.
Sistemas de pruebas multivariantes: Los sistemas de pruebas multivariantes se desarrollaron para investigar diferentes especificidades de autoanticuerpos en un solo paso y ahorrar tiempo en comparación con el procedimiento de varios pasos descrito anteriormente. Uno es un ELISA en el que las proteínas recombinantes se colocaron en una única placa ELISA [18], el otro se basa en la tecnología Biochip®. Aquí, hasta 12 sustratos que miden sólo aprox. 1×1 mm se compilan en un campo de incubación sobre un portaobjetos normal de laboratorio y la unión del autoanticuerpo se hace visible mediante IF indirecta. Además de sustratos de órganos como el esófago de primate y la piel humana dividida, también están disponibles BP180 NC16A recombinante y Dsg1, Dsg3, BP230, laminina 332, colágeno tipo VII expresado en la superficie celular de células HEK293 [19].
Inmunoblot e inmunoprecipitación
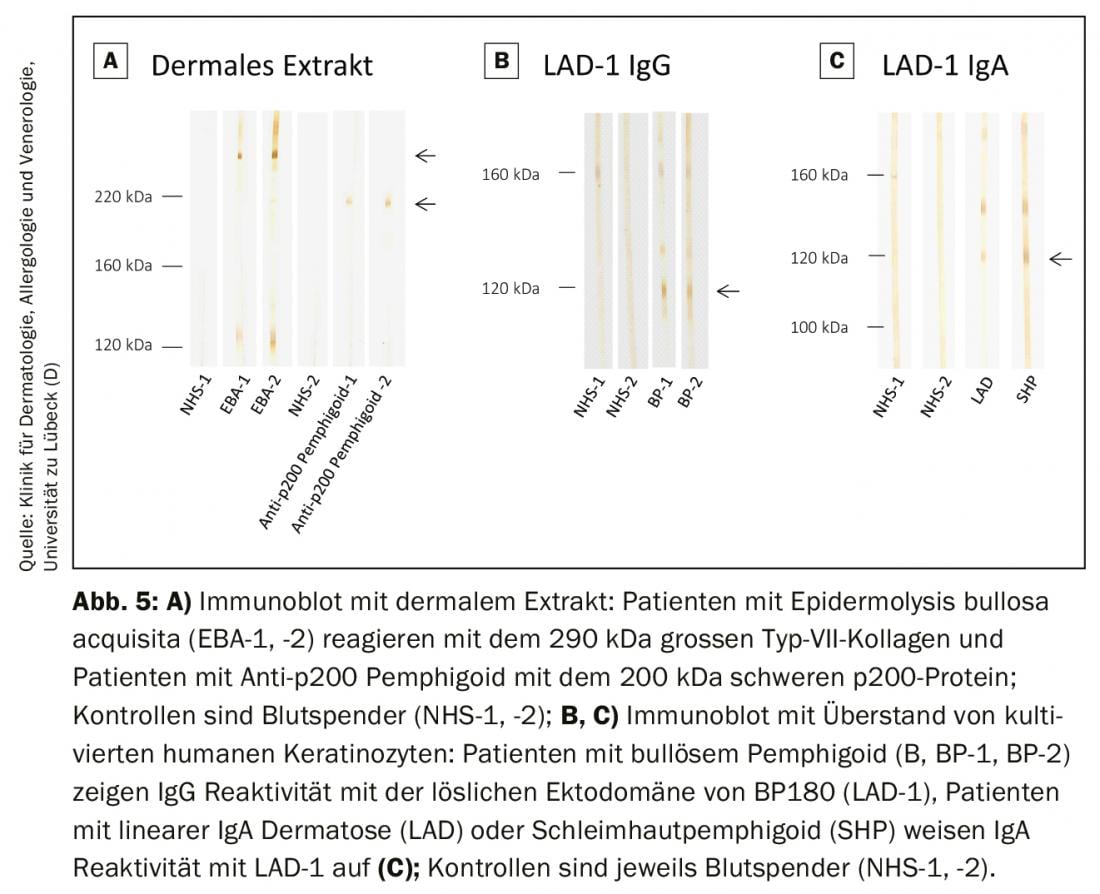
En los laboratorios especializados, estos métodos se ofrecen como procedimientos internos para detectar aquellas especificidades de autoanticuerpos que aún no están cubiertas por los sistemas de pruebas comerciales. Esto incluye la detección de autoanticuerpos contra la molécula completa de colágeno de tipo VII, así como contra la proteína p200. (Fig. 5A) y la detección de anticuerpos IgG e IgA contra el ectodominio del BP180, que es la región inmunodominante en el penfigoide mucoso y las dermatosis IgA lineales. (Fig. 5B y C). Los anticuerpos contra la periplakina, la desmoplaquina I/II, la plectina, la epiplakina, las desmocolinas y la α2-macroglobulina-like 1 en el pénfigo paraneoplásico pueden visualizarse en diversos análisis de inmunoblot e inmunoprecipitación. En Alemania, el organismo alemán de acreditación (DAkkS) ofrece la posibilidad de auditar externamente estos procedimientos internos para garantizar el mayor nivel posible de diagnóstico. El laboratorio autoinmune de la Clínica Universitaria de la Piel de Lübeck ofrece estos procedimientos (DAkkS D-ML-13069-06-00; www.uksh.de/dermatologie-luebeck).
Constelaciones especiales de hallazgos
En caso de constelaciones poco claras de hallazgos, se recomienda volver a tomar una biopsia de muestra perilesional para IF directa [4]. En el penfigoide de las mucosas, se demostró que en el 16% de los pacientes el diagnóstico sólo podía confirmarse mediante una segunda IF directa [21]. La IF directa es especialmente importante en el penfigoide mucoso y la epidermólisis bullosa adquirida, ya que no pueden detectarse autoanticuerpos circulantes en el 50% de los casos.
En la gran mayoría de los pacientes con BAID, el diagnóstico ya puede realizarse hoy en día serológicamente con la ayuda de sistemas de pruebas comerciales si el cuadro clínico es compatible [18,19]. También recomendamos las pruebas serológicas como examen inicial de detección si se va a prescindir inicialmente de una biopsia de muestra, por ejemplo, en niños o pacientes >75 años con >6 semanas de prurito existente que no pueda explicarse por otra enfermedad.
La directriz actual de la AWMF [5] recomienda el diagnóstico de pénfigo vulgar/foliáceo en caso de
- Cuadro clínico adecuado e inmunofluorescencia directa positiva
- Coincidencia de cuadro clínico y reactividad con desmogleína 1 ó 3 en ELISA/en células transfectadas.
- Cuadro clínico e histopatología coincidentes e inmunofluorescencia indirecta positiva en secciones esofágicas de mono.
Diagnóstico de penfigoide bulloso en caso de
- cuadro clínico apropiado y IF directa positiva y reactividad con BP180 y/o BP 230
- Cuadro clínico adecuado y IF directa positiva y unión epidérmica de IgG en la IF indirecta en piel escindida.
- Cuadro clínico con ampollas abultadas y unión epidérmica de IgG en IF indirecta sobre piel dividida o secciones de esófago de mono y reactividad con BP180 y/o BP230.
- Cuadro clínico con ampollas abultadas e histopatología coincidente y unión epidérmica de IgG en IF indirecta en piel escindida.
- cuadro clínico e histopatología coincidentes (escisión subepidérmica) y reactividad con BP180
- Cuadro clínico con burbujas abultadas y reactividad clara con BP180 (por ejemplo, >3 veces el límite inferior de detección en ELISA comercial).
Resumen
Las dermatosis ampollosas autoinmunes (BAID) son un grupo de enfermedades raras, potencialmente mortales, que se manifiestan clínicamente como lesiones en la piel o las mucosas y no siempre tienen por qué estar asociadas a la formación de ampollas. Las alteraciones de la piel y las mucosas se desencadenan por una reacción del sistema inmunitario mediada por autoanticuerpos contra proteínas estructurales de la piel que provocan la integridad del propio epitelio (desmosomas) o la del anclaje epitelial al tejido conectivo subyacente de la piel o las mucosas. Las primeras se agrupan como enfermedades del pénfigo y las segundas como enfermedades del penfigoide. Debe distinguirse de la dermatitis herpetiforme, en la que se forman anticuerpos contra la transglutaminasa 2 y 3 y que siempre va acompañada de enfermedad celíaca. El antígeno diana de los autoanticuerpos tiene una influencia decisiva en la patogénesis y, por tanto, en el aspecto clínico de la BAID, y su determinación no sólo sirve para establecer un diagnóstico exacto, sino que también influye decisivamente en el pronóstico y la terapia a aplicar. El diagnóstico de la BAID se basa en la detección de autoanticuerpos unidos al tejido en la inmunofluorescencia directa de una biopsia de muestra perilesional y en la detección de autoanticuerpos circulantes. Conociendo el cuadro clínico, las modernas pruebas ELISA y de inmunofluorescencia indirecta basadas en las regiones immmundominantes de los antígenos diana ya pueden diagnosticar serológicamente al 80-90% de los pacientes con BAID. El desarrollo de otros sistemas de pruebas estandarizados, sensibles y específicos, simplificará aún más el diagnóstico de la BAID.
Mensajes para llevarse a casa
- Las dermatosis autoinmunes bullosas son enfermedades raras desencadenadas por autoanticuerpos contra proteínas estructurales de la piel.
- Con diferencia, la dermatosis bullosa autoinmune más común en el norte y centro de Europa es el penfigoide bulloso, que se caracteriza clínicamente por una edad avanzada, prurito grave, ampollas abultadas y asociación con enfermedad neurológica.
- El diagnóstico exacto tiene relevancia pronóstica y terapéutica.
- Existen pruebas ELISA e inmunofluorescencia indirecta estandarizadas, altamente sensibles y específicas, que utilizan la región inmunodominante de los antígenos diana.
- Conociendo el cuadro clínico, las pruebas serológicas permiten un diagnóstico fiable en la gran mayoría de los pacientes; la biopsia de muestra perilesional para el examen de inmunofluorescencia directa sigue siendo el patrón de oro diagnóstico.
Agradecimiento
Nos gustaría dar las gracias a Ingeborg Atefi y Marina Kongsbak-Reim por su apoyo con las imágenes de fluorescencia y a Vanessa Krull por su ayuda con los inmunoblots. El trabajo contó con financiación estructural del Cluster de Excelencia 2167/1 Medicina de Precisión en Inflamación Crónica.
Literatura:
- Schmidt E, Kasperkiewicz M, Joly P: Pénfigo. Lancet 2019; 394: 882-894.
- Schmidt E, Zillikens D: Enfermedades penfigoides. Lancet 2013; 381: 320-332.
- van Beek N, Zillikens D, Schmidt E: Diagnóstico de las enfermedades ampollosas autoinmunes. J Dtsch Dermatol Ges 2018; 16: 1077-1091.
- Schmidt E, Goebeler M, Hertl M, et al: Directriz S2k para el diagnóstico del pénfigo vulgar/foliáceo y el penfigoide bulloso. J Dtsch Dermatol Ges 2015; 13: 713-727.
- Schmidt E, Sticherling M, Sardy M, et al: Directrices S2k para el tratamiento del pénfigo vulgar/foliáceo y el penfigoide bulloso: actualización de 2019. J Dtsch Dermatol Ges 2020; 18: 516-526.
- Hübner F, Recke A, Zillikens D, et al.: Prevalencia y distribución por edades de las enfermedades penfigicas y penfigoides en Alemania. Revista de dermatología investigadora (JEADV) 2016: 136(12): 2495-2498; doi: 10.1016/j.jid.2016.07.013.
- Hübner F, König IR, Holtsche MM, et al.: Prevalencia y distribución por edades del pénfigo y las enfermedades penfigoides entre los pacientes pediátricos en Alemania. En: JEADV 2020; doi: 10.1111/jdv.16467.
- Marazza G, Pham HC, Scharer L, et al: Incidencia del penfigoide bulloso y el pénfigo en Suiza: un estudio prospectivo de 2 años. Br J Dermatol 2009; 161: 861-868.
- Sadik CD, Schmidt E: Resolución en el penfigoide bulloso. Seminarios de inmunopatología 2019; 41: 645-654.
- della Torre R, Combescure C, Cortes B, et al: Presentación clínica y retraso diagnóstico en el penfigoide bulloso: una cohorte prospectiva a escala nacional. Br J Dermatol 2012; 167: 1111-1117.
- Schulze F, Neumann K, Recke A, et al: Malignidades en las enfermedades penfígicas y penfigoides. J Invest Dermatol 2015; 135: 1445-1447.
- Kridin K, Cohen AD: Penfigoide ampolloso asociado a inhibidores de la dipeptidil-peptidasa IV: Una revisión sistemática y un metaanálisis. J Am Acad Dermatol 2018.
- Goletz S, Hashimoto T, Zillikens D, Schmidt E: Penfigoide anti-p200. J Am Acad Dermatol 2014; 71: 185-191.
- Vorobyev A, Ludwig RJ, Schmidt E: Características clínicas y diagnóstico de la epidermólisis ampollosa adquirida. Expert Rev Clin Immunol 2017; 13: 157-169.
- Kasperkiewicz M, Ellebrecht CT, Takahashi H, et al: Pénfigo. Nature reviews Primers de enfermedades 2017; 3: 17026.
- Egan CA, Lazarova Z, Darling TN, et al: Penfigoide cicatricial antiepiligrina y riesgo relativo de cáncer. Lancet 2001; 357: 1850-1851.
- Goletz S, Probst C, Komorowski L, et al: Un ensayo sensible y específico para el diagnóstico serológico del penfigoide de mucosas antilaminina 332. Br J Dermatol 2019; 180: 149-156.
- van Beek N, Dahnrich C, Johannsen N, et al: Estudios prospectivos sobre el uso rutinario de un nuevo ensayo inmunoenzimático multivariante para el diagnóstico de las enfermedades ampollosas autoinmunes. J Am Acad Dermatol 2017; 76: 889-894 e5.
- van Beek N, Kruger S, Fuhrmann T, et al: Estudio prospectivo multicéntrico sobre el diagnóstico multivariante de las dermatosis ampollosas autoinmunes mediante la tecnología BIOCHIP(TM). J Am Acad Dermatol 2020.
- van Beek N, Rentzsch K, Probst C, et al: Diagnóstico serológico de las enfermedades cutáneas ampollosas autoinmunes: Comparación prospectiva de la técnica de inmunofluorescencia indirecta basada en el mosaico BIOCHIP con la estrategia convencional de prueba única en varios pasos. Orphanet J Rare Dis 2012; 7: 49.
- Shimanovich I, Nitz JM, Zillikens D: El muestreo múltiple y repetido aumenta la sensibilidad de las pruebas de inmunofluorescencia directa para el diagnóstico del penfigoide de las mucosas. J Am Acad Dermatol 2017; 77: 700-705 e3.
PRÁCTICA DERMATOLÓGICA 2020; 30(5): 12-19