El síndrome, descrito por primera vez por los doctores John Crigler y Victor Najjar en 1952, se caracteriza por un trastorno congénito en el metabolismo de la bilirrubina. Las opciones de tratamiento existentes pretenden reducir la cantidad de bilirrubina no conjugada en la sangre. Sin embargo, esto requiere a veces procedimientos relativamente complejos. Las alternativas incluyen el trasplante de hígado o posiblemente un trasplante de hepatocitos. Pero hay luz en el horizonte: una terapia génica ha logrado resultados prometedores en un ensayo clínico.
La bilirrubina es un producto de degradación del pigmento rojo de la sangre, la hemoglobina, y se forma cuando los glóbulos rojos se descomponen. [1–3]Normalmente, la enzima UGT1A1 (UDP-glucuronosiltransferasa 1 polipéptido A1) cataliza la formación del diglucurónido de bilirrubina hidrosoluble en el retículo endoplásmico liso del hígado, que luego se excreta al intestino a través de los conductos biliares . Sin embargo, las personas afectadas por el síndrome de Crigler-Najjar carecen de esta enzima, lo que hace que la bilirrubina se acumule en el organismo y, sin tratamiento, provoque daños neurológicos importantes o incluso la muerte [1]. En el síndrome de Crigler-Najjar tipo 1, la enzima UGT está completamente inactiva y en el tipo 2 está muy reducida [4]. Ambas formas están causadas por defectos genéticos en el gen UGT1A1 del cromosoma 2. Debido a la naturaleza genética de la enfermedad, ambos progenitores deben ser portadores de la mutación para que su hijo se vea afectado [2]. Se calcula que menos de 1 de cada 1 millón de recién nacidos en todo el mundo están afectados por el síndrome de Crigler-Najjar.
Aspecto clínico
El síndrome de Crigler-Najjar tipo 1 suele manifestarse inmediatamente después del nacimiento como una hiperbilirrubinemia excesiva que, si no se trata, suele desembocar en un kernicterus con graves daños neurológicos. Como consecuencia, los pacientes afectados suelen morir en la primera infancia si no reciben tratamiento. [11]Los recién nacidos son especialmente susceptibles a los daños neurológicos causados por la hiperbilirrubinemia, ya que el hígado, aún en desarrollo, está sometido a un gran estrés por la descomposición de la hemoglobina fetal en los primeros días de vida y la barrera hematoencefálica aún no está muy bien desarrollada . Los niveles séricos de bilirrubina ligeramente elevados y la ictericia no son infrecuentes en los recién nacidos, pero los niveles de bilirrubina deben vigilarse cuidadosamente si hay algún signo de aumento.
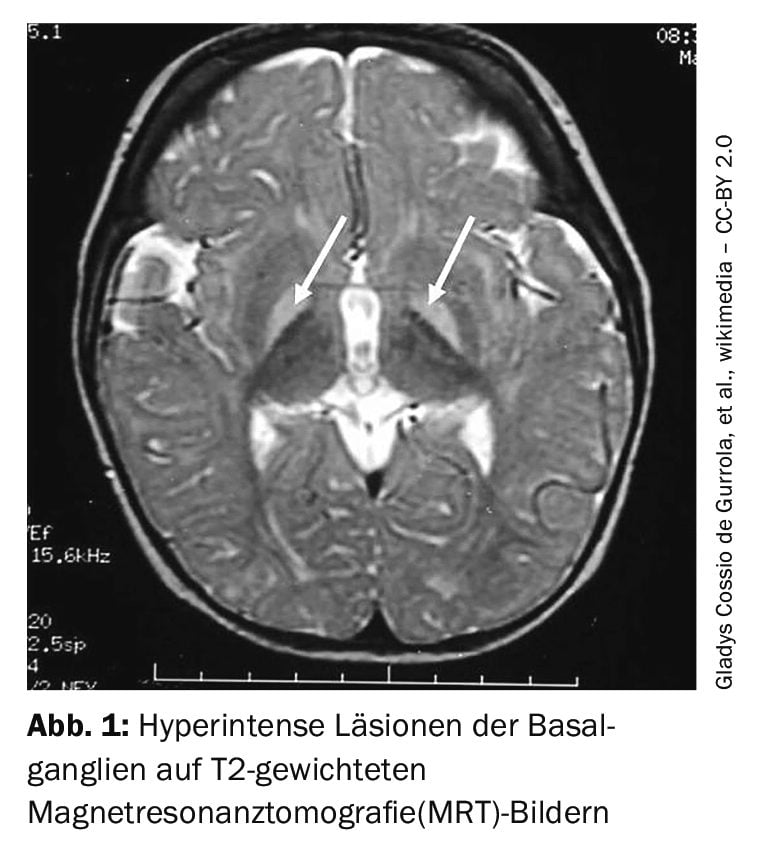
El síndrome de Crigler-Najjar tipo 2 es menos grave que el tipo 1, en el que el kernicterus es poco frecuente, pero los síntomas perturbadores con coloración amarillenta de la piel y prurito extenso pueden perjudicar gravemente la calidad de vida [4]. En algunas personas, el diagnóstico no se realiza hasta la edad adulta, como en el informe de caso resumido en el recuadro [5]. El kernicterus es poco frecuente en el tipo 2, pero puede aparecer sobre todo cuando la persona afectada está enferma, no come o está anestesiada [2]. La posibilidad de que una ictericia grave a los pocos días de nacer sea el síndrome de Crigler-Najjar puede confirmarse mediante la evaluación clínica, los antecedentes familiares y las pruebas genéticas y de laboratorio. Un hallazgo clásico sería, por ejemplo, un nivel elevado de bilirrubina no conjugada en la sangre o una falta de bilirrubina conjugada en la bilis. Las pruebas genéticas para identificar mutaciones en el gen UGT1A1 pueden confirmar el diagnóstico [2].
Informe de un caso: diagnóstico de SNC tipo 2 en la edad adulta Un hombre de 21 años había padecido episodios recurrentes de ictericia desde la primera infancia. Durante los últimos 6 meses, había estado especialmente preocupado por una ictericia persistente acompañada de vómitos ocasionales. |
| Historia: Su nacimiento transcurrió sin complicaciones en su momento, no hubo ictericia neonatal ni necesidad de transfusiones de sangre. Alcanzó los hitos de desarrollo esperados para su edad y no hubo motivo de preocupación. A los 5 años, sus padres notaron por primera vez una extraña decoloración amarillenta de los ojos, que no se acompañaba de fiebre, picor, dolor abdominal ni heces de color arcilla, pero la orina estaba muy decolorada. El paciente se sometió entonces a diversos tratamientos alternativos y complementarios. Desgraciadamente, ninguna de estas medidas condujo a la curación completa de su afección. Según recuerdan los padres de la paciente, el nivel más alto de bilirrubina sérica que se le midió fue de 12 mg/dL. |
| Investigaciones diagnósticas actuales: El examen actual reveló ictericia sin organomegalia. Otras investigaciones clínicas también mostraron resultados normales y no ofrecieron ninguna explicación inmediata para su ictericia persistente. Sin embargo, las investigaciones rutinarias revelaron hiperbilirrubinemia indirecta con niveles normales de enzimas hepáticas. El examen ecográfico del abdomen no presentaba observaciones. A continuación se realizó un examen hemolítico completo, pero todas las pruebas fueron negativas. Basándose en el inicio, el curso y la presencia de hiperbilirrubinemia no conjugada, se hizo un diagnóstico provisional de hiperbilirrubinemia no conjugada no hemolítica. Ante la sospecha de un síndrome de bilirrubinemia indirecta congénita, se inició un análisis en busca de mutaciones de UGT1A1, que resultó positivo e indicó la presencia de una deficiencia enzimática parcial. Esto condujo al diagnóstico definitivo de síndrome de Crigler-Najjar tipo 2. |
| Terapia: Para controlar los síntomas del paciente y mejorar su calidad de vida, se le administró fenobarbital oral a una dosis de 5 mg/kg. Sorprendentemente, se observó una reducción significativa de los niveles séricos de bilirrubina del paciente a las dos semanas de iniciar el tratamiento. Así pues, la terapia demostró ser eficaz. |
| según [5] |
Opciones terapéuticas disponibles actualmente
El objetivo principal del tratamiento de los pacientes con síndrome de Crigler-Najjar es reducir la cantidad de bilirrubina no conjugada en la sangre de la forma más rápida y constante posible. Esto se recomienda para el síndrome de Crigler-Najjar tipo 1 (SNC I) y el tipo 2 (SNC II) de diferentes maneras [2].
El tratamiento conservador del SNC I se basa en tres pilares [4]:
- Fototerapia diaria constante con luz azul (esto hace que la bilirrubina sea hidrosoluble)
- Administración de tinprotoporfirina, un inhibidor de la hemooxigenasa (atenúa los aumentos de bilirrubina)
- Administración de carbonato cálcico y fosfato cálcico (aumenta la secreción de bilirrubina no conjugada en el intestino)
Esta terapia puede prolongar la esperanza de vida y retrasar la aparición de complicaciones neurológicas. Otra opción terapéutica, el trasplante de hígado, debe intentarse lo antes posible. El trasplante alogénico de hepatocitos se encuentra actualmente en fase experimental.
El SNC II se trata administrando fenobarbital una vez al día [4]. Otra alternativa es la rifampicina. Al inducir la actividad enzimática, la concentración de bilirrubina en el plasma puede reducirse a niveles seguros.
Estudio de terapia génica: oportunidades y riesgos D’Antiga et al. [6,7]investigaron la seguridad y eficacia de una única infusión intravenosa de un vector AAV que codifica UGT1A1 en 5 pacientes con síndrome de Crigler-Najjar . |
| En tres de los tratados con una dosis más alta, los niveles de bilirrubina cayeron por debajo de 30 µmol por litro (17,5 mg/dl), lo que significó que se pudo interrumpir la fototerapia durante los siguientes 18 meses de seguimiento [7]. Sin embargo, no se consiguió la normalización completa del nivel de bilirrubina en ningún caso. |
| Según Di Dato et al. [6,8]la duración de la eficacia de una única infusión del vector AAV sigue sin estar clara en la actualidad . Además, se ha informado de que, en pacientes con hemofilia, la terapia génica puede conducir al desarrollo de anticuerpos AAV neutralizantes persistentes, de alto título y de reactividad cruzada, lo que podría descartar la posibilidad de nuevas administraciones del vector [9]. [10]Las infusiones múltiples con vectores AAV también pueden suponer un riesgo de genotoxicidad . |
La terapia génica como posible método de tratamiento alternativo
El uso de vectores AAV (virus adeno-asociados), por ejemplo, ha sido aprobado por la Administración de Alimentos y Medicamentos de Estados Unidos (FDA) para la terapia de sustitución génica en pacientes con atrofia muscular espinal y ceguera congénita. [1,7]Los resultados iniciales de un ensayo clínico sobre el síndrome de Crigler-Najjar sugieren que la terapia génica basada en AAV podría servir como posible tratamiento alternativo para esta enfermedad potencialmente mortal . El tratamiento, actualmente en fase de ensayo, ha sido desarrollado por investigadores del Généthon**. Consiste en proporcionar a las células hepáticas una copia del gen UGT1A1, que codifica una enzima que facilita la eliminación de la bilirrubina. Las observaciones iniciales del estudio CureCN (“Terapia génica hepática mediada por vectores de virus adenoasociados para el síndrome de Crigler-Najjar”) sugieren que la terapia génica podría ser un tratamiento alternativo potencial. [Adeno-assoziiertes Virus] “Estamos muy entusiasmados con los resultados obtenidos hasta ahora en este ensayo de terapia génica mediada por AAV para el tratamiento del síndrome de Crigler-Najjar”, señaló el Dr. D’Antiga [1]. “El tratamiento ha demostrado ser seguro a dosis adecuadas y capaz de dirigirse a la enfermedad hasta tal punto que la primera paciente pudo interrumpir su fototerapia diaria, eliminando el riesgo de daños neurológicos. El grado de mejora de la segunda paciente sugiere que ella también podrá dejar pronto la fototerapia” [1].
** Genethon forma parte del Instituto de Bioterapias para Enfermedades Raras (BIRD)
Un artículo publicado en 2024 por Di Dato et al. [6] también evalúa la terapia génica como una posible alternativa terapéutica prometedora, aunque los autores señalan que aún quedan algunas preguntas sin respuesta sobre la eficacia y la seguridad de este enfoque terapéutico, que son objeto de investigaciones en curso (recuadro).
Literatura:
- “Un estudio clínico da esperanzas a las personas que padecen una rara enfermedad genética del hígado”, https://cordis.europa.eu/article/id/430456-clinical-trial-gives-hope-to-sufferers-of-rare-genetic-liver-disease/de,(última consulta: 29/08/2024).
- “Síndrome de Crigler-Najjar”, https://liverfoundation.org,(último acceso 29/08/2024).
- “Mutación de la UDP-glucuroniltransferasa (UGT1A1*28)”,www.labor-duesseldorf.de/examination/view/udp-glukuronyltransferase-mutation-ugt1a128,(última consulta: 29/08/2024).
- “Síndrome de Crigler-Najjar”, https://flexikon.doccheck.com,(último acceso 29/08/2024).
- Rijal D, et al: Un caso poco frecuente de síndrome de Crigler-Najjar tipo 2: Informe de un caso y revisión de la literatura. Clin Case Rep 2023; 13 de noviembre; 11(11): e8176.
- Di Dato F, D’Uonno G, Iorio R: Síndrome de Crigler-Najjar: mirar al futuro no hace olvidar el presente. Orphanet J Rare Dis. 2024 mar 7; 19(1): 102.
- D’Antiga L, et al: Terapia génica en pacientes con el síndrome de Crigler-Najjar. NEJM 2023; 389(7): 620-631.
- Aronson SJ, Ronzitti G, Bosma PJ: ¿Qué es lo próximo en terapia génica para el síndrome de Crigler-Najjar? Expert Opin Biol Ther 2023; 23(2): 119-121.
- George LA, et al: Long-term Follow-Up of the First in Human Intravascular Delivery of AAV for Gene transfer: AAV2-hFIX16 for severe Hemophilia B. Mol Ther 2020; 28(9): 2073-2082.
- Sabatino DE, et al: Evaluación del estado de la ciencia para la integración del virus adeno-asociado: una perspectiva integrada. Mol Ther 2022; 30(8): 2646-2663.
- Wikipedia: Kernicterus, https://en.wikipedia.org,(última consulta: 29/08/2024).
PRÁCTICA GP 2024; 19(9): 44-45