
Este síndrome muy poco frecuente está causado por mutaciones en el gen LRP2, que codifica la megalina, y se hereda de forma autosómica recesiva. Se caracteriza por rasgos craneofaciales con rasgos faciales característicos. Además de las complicaciones oculares y la pérdida auditiva neurosensorial, se describen otras anomalías clínicas. La sospecha diagnóstica puede confirmarse mediante pruebas genéticas. Dependiendo de la evolución individual, se requieren diversas medidas terapéuticas.
El síndrome de Donnai-Barrow (SDB) está asociado a múltiples malformaciones congénitas [1]. Los individuos afectados se caracterizan por una dismorfia facial típica, miopía y otros hallazgos oculares, así como pérdida de audición, agenesia del cuerpo calloso, proteinuria de bajo peso molecular y diversos trastornos del desarrollo intelectual. Son frecuentes la hernia diafragmática congénita y/o el onfalocele. Se dispone de pocos datos sobre la prevalencia y la incidencia de la ECP. Hasta la fecha, se han descrito menos de 50 individuos afectados de unas 20 familias. La ECP se da en todos los grupos étnicos y no parece existir un gradiente de género. La ECP está causada por mutaciones en el gen LRP2 (proteína 2 relacionada con el receptor de lipoproteínas de baja densidad; 2q31.1). Este gen codifica la megalina, que se expresa en varios epitelios reabsorbentes, sobre todo en el cerebro, los riñones y los ojos. La megalina desempeña un papel importante en varias cascadas de señalización y en la endocitosis de muchos ligandos.
Características clínicas comunes
Casi todos los pacientes presentan los siguientes síntomas [1]:
- Agenesia/hipogénesis del cuerpo calloso
- Fontanela anterior agrandada
- Hipoacusia neurosensorial pronunciada (es decir, el sonido llega al oído interno pero no puede convertirse en impulsos nerviosos o éstos no se transmiten al cerebro).
- Hipertelorismo.
Los rasgos faciales característicos son [1]:
- fisuras palpebrales inclinadas hacia abajo,
- Nariz corta con un puente nasal plano,
- Frente alta y ancha
- “pico de viuda” en la línea frontal del cabello y a veces proptosis
Alrededor del 40% de los pacientes presentan una hernia diafragmática congénita y/o un onfalocele. El desarrollo suele retrasarse y la inteligencia se reduce en diversos grados. La miopía elevada (>6 dptr) puede provocar desprendimiento/distrofia retiniana y una discapacidad visual creciente. Ocasionalmente, se ha informado de coloboma del iris, glomeruloesclerosis segmentaria focal y disfunción de los túbulos proximales (que rara vez conduce a insuficiencia renal).
Diagnóstico y DD El diagnóstico del síndrome de Donnai-Barrow (SDB) se realiza por una combinación de características clínicas y de imagen junto con un patrón típico de proteinuria de bajo peso molecular, niveles elevados en orina de proteína de unión al retinol (RBP) y cociente RBP/creatinina. El diagnóstico se confirma mediante un análisis de ADN. Debido a la constelación característica de síntomas, el número de diagnósticos diferenciales de la ECP es limitado. Algunos síntomas comunes incluyen la tetrasomía 12p, así como los síndromes de Fryns, Chudley-McCullough, acrocallosal y cráneo-fronto-nasal. El fenotipo renal es en parte similar a la enfermedad de Dent y al síndrome de Lowe. El fenotipo ocular puede indicar el síndrome de Stickler. La detección de hipertelorismo y hernia diafragmática congénita u onfalocele en el diagnóstico prenatal por imagen debe sugerir la ECP. El diagnóstico prenatal en embarazos de alto riesgo requiere la identificación previa de las mutaciones causantes de la enfermedad en la familia. |
| según [1] |
Manejo terapéutico y pronóstico
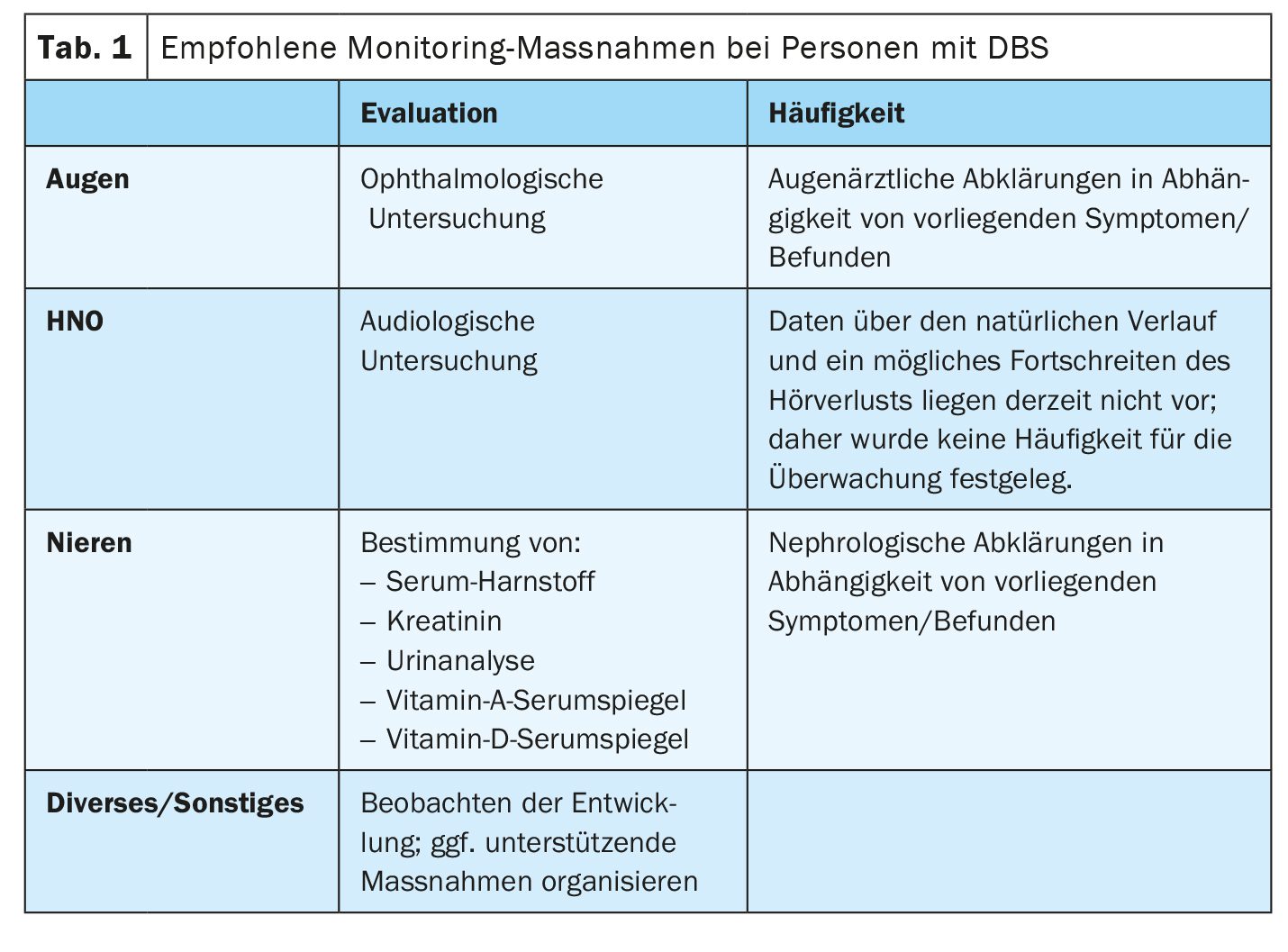
La ECP se hereda de forma autosómica recesiva. Debe ofrecerse asesoramiento genético a los padres de los niños afectados y a sus familiares [1]. A excepción de un único caso publicado de disomía uniparental, los padres de los pacientes cuyos casos se documentaron resultaron ser heterocigotos obligados. Es necesario realizar revisiones periódicas de la agudeza visual, la audición y la función renal. La corrección de las gafas, el tratamiento para prevenir el desprendimiento de retina, los audífonos y/o los implantes cocleares forman parte del plan de tratamiento. La hernia diafragmática congénita y/o el onfalocele pueden requerir una intervención quirúrgica. Deben ofrecerse medidas de apoyo específicas a los niños afectados. Los afectados pueden conseguir una visión y una audición utilizables con corrección. El estado general de salud de los pacientes en la infancia y la adolescencia suele ser bueno. La insuficiencia renal terminal es una complicación rara y potencialmente mortal. La presentación pre o perinatal con defectos diafragmáticos y de la pared abdominal requiere intervención quirúrgica y se asocia a una mayor morbilidad y mortalidad.
Estudio de caso: progresión desde el nacimiento hasta la edad escolar primaria
Un niño que actualmente tiene 9 años nació de padres caucásicos sanos y no emparentados que en ese momento tenían 34 años (la madre) y 40 años (el padre) [2]. Tenía una hermana sana y dos hermanastros sanos por parte de madre. Durante el embarazo, se detectó un pequeño exomphalos mediante ecografía. La paciente nació por parto vaginal normal. Los exámenes clínicos y de imagen postnatales revelaron hipertelorismo pronunciado, coloboma bilateral (formación de hendiduras en la zona ocular), ausencia del cuerpo calloso, malrotación del intestino, hernias inguinales bilaterales, pero ninguna hernia diafragmática congénita.
Onfalocele: La hernia umbilical se redujo el primer día, sus hernias se repararon al año de edad y la malrotación se operó finalmente a los 18 meses.
Agenesia del cuerpo calloso: A la edad de 4 meses, el perímetro craneal de la paciente era de 44,5 cm (percentil 95), la altura de 60,6 cm (percentil 90) y el peso de 5,78 kg (percentil 90). La resonancia magnética del cerebro confirmó la agenesia del cuerpo calloso y también mostró un encefalocele frontal con una fosa anterior ensanchada y una malformación de Chiari 1 con amígdalas cerebelosas que se extendían hasta C1.
Manifestaciones oculares: Además de colobomas bilaterales del iris y coriorretinianos, presentaba una miopía elevada, así como una catarata inferior derecha y un lenticono posterior izquierdo, que le habían sido diagnosticados a los 3 meses de edad. La miopía estaba asociada a globos oculares agrandados (longitud axial de 30 mm a los 7 años de edad) y estafilomas posteriores bilaterales, y se le practicó una retinopexia profiláctica con láser de 360 grados para prevenir el desprendimiento de retina. Su graduación de gafas era OD (Oculus Dexter) -15,00 D, OS (Oculus Sinister) -19,25/-2,00 eje 92°, aunque normalmente llevaba lentes de contacto y alcanzaba una agudeza visual corregida de OD 20/200, OS 20/100. A la edad de 7 años, la paciente presentaba unos resultados electrodiagnósticos normales, pero refirió un deterioro visual en los últimos 2 años, especialmente por la noche. Una electrorretinografía repetida a la edad de 9 años reveló una disfunción retiniana generalizada que afectaba tanto al sistema de bastones como al de conos, principalmente en la interfase entre el fotorreceptor y el epitelio pigmentario de la retina. Las masas oculares a los 6 años de edad eran de 45, 70 y 110 mm para la distancia cantal interna, pupilar y cantal externa, respectivamente.
Hallazgos audiológicos: Debido a una sordera bilateral grave, a la paciente se le colocaron implantes cocleares a los 4 años, que se revisaron a los 6 años y de nuevo a los 8 años.
Desarrollo escolar: El paciente asiste a una escuela ordinaria y recibe apoyo especial para sus déficits visuales y auditivos. Presenta cierto retraso en su desarrollo y asiste a una clase en la que va dos años por detrás de sus compañeros. Sin embargo, progresa adecuadamente en esta clase y se supone que gran parte de su retraso en el desarrollo se debe a su deficiencia bisensorial y a las lagunas en su escolarización debidas a las frecuentes hospitalizaciones.
Confirmación del diagnóstico: La sospecha diagnóstica de ECP se confirmó mediante pruebas genéticas, en las que se detectó por secuenciación directa una deleción homocigota de 4 pb (c.11469_11472delTTTG) en el exón 60 del gen LRP2.
Literatura:
- “Síndrome de Donnai-Barrow”, www.orpha.net,(última consulta: 27/09/2024).
- Kantarci S, et al: Síndrome de Donnai-Barrow (SDB/FOAR) en un niño con una mutación homocigota de LRP2 debida a una isodisomía paterna completa del cromosoma 2. Am J Med Genet A 2008; 146A(14): 1842-1847.
- Longoni M, et al: Síndrome de Donnai-Barrow. [Updated 2018 Nov 21] 2008 Ago 28 . En: Adam MP, et al. (Eds). [Internet]GeneReviews® . Seattle (WA): Universidad de Washington, Seattle; 1993-2024.
PRÁCTICA GP 2024; 19(10): 24-25









