
El síndrome de Fanconi-Bickel (FBS) se produce debido a variantes en el gen SLC2A2. El diagnóstico de una enfermedad genética rara puede tardar hasta 5-6 años, e incluso más en países de ingresos bajos y medios con recursos tecnológicos limitados. Médicos de Perú presentaron el caso de un niño de dos años y medio con retraso del crecimiento, hepatomegalia, acidosis metabólica, hipofosfatemia, hipopotasemia e hiperlactatemia.
El síndrome de Fanconi-Bickel (OMIM #227810), un trastorno hereditario autosómico recesivo (RA), se caracteriza por una combinación de enfermedad hepática y renal causada por un defecto en el transportador de glucosa GLUT2 (gen SLC2A2). Esto conduce a la acumulación de glucógeno, la disfunción tubular renal proximal y el deterioro de la utilización de la glucosa y la galactosa.
El fenotipo incluye falta de aumento de peso, distensión abdominal, hepatomegalia, hipoglucemia en ayunas, hiperglucemia posprandial, glucosuria, fosfaturia, aminoaciduria, poliuria, acidosis metabólica, osteoporosis, hipofosfatemia, raquitismo y presencia de glucógeno en la biopsia hepática o renal. En raras ocasiones, se ha observado un carcinoma hepatocelular debido a la activación de la vía de señalización Wnt. Sin embargo, también se han descrito casos de pacientes con síntomas clínicos leves, incluidos aquellos con glucosuria pura. El gen SLC2A2 (OMIM *138160) contiene 11 exones, y su proteína GLUT2 consta de 524 aminoácidos y se localiza en la membrana celular, expresándose en hepatocitos, enterocitos, túbulos proximales renales, células beta pancreáticas, neuronas y astrocitos. Las variantes patogénicas del SLC2A2 alteran la entrada y salida de glucosa en los hepatocitos y reducen la secreción de insulina debido a una mayor sensibilidad de las células beta en la fase posprandial.
Caso clínico de un niño de 2½ años
Un niño de 2 años y 7 meses, nacido y criado en Perú del quinto embarazo de padres emparentados, se presentó al equipo del Dr. Hernán Abarca-Barriga del Instituto Nacional de Salud (INS) Perú debido a vómitos, diarrea, acidosis metabólica, hipopotasemia e hiperlactatemia, hipoactividad y fiebre. Debido a estos síntomas, había sido hospitalizado previamente a la edad de 1 año y 10 meses y 2 años y 2 meses [1].
La niña nació con un peso al nacer de 3620 g, una altura de 49 cm y un perímetro cefálico de 34 cm (percentil normal) y una puntuación Apgar de 8-9. En cuanto al desarrollo psicomotor, consiguió controlar la cabeza al mes, se sentó sin ayuda a los siete meses y caminó con apoyo al año y seis meses. Pronunció sus primeras palabras al año y cinco meses, dijo dos palabras a los dos años y nueve meses y mostró una sonrisa social al año. Al año y ocho meses de edad, se le evaluó por bajo peso y crecimiento reducido, diarrea crónica, fiebre y aumento del volumen abdominal (Fig. 1).
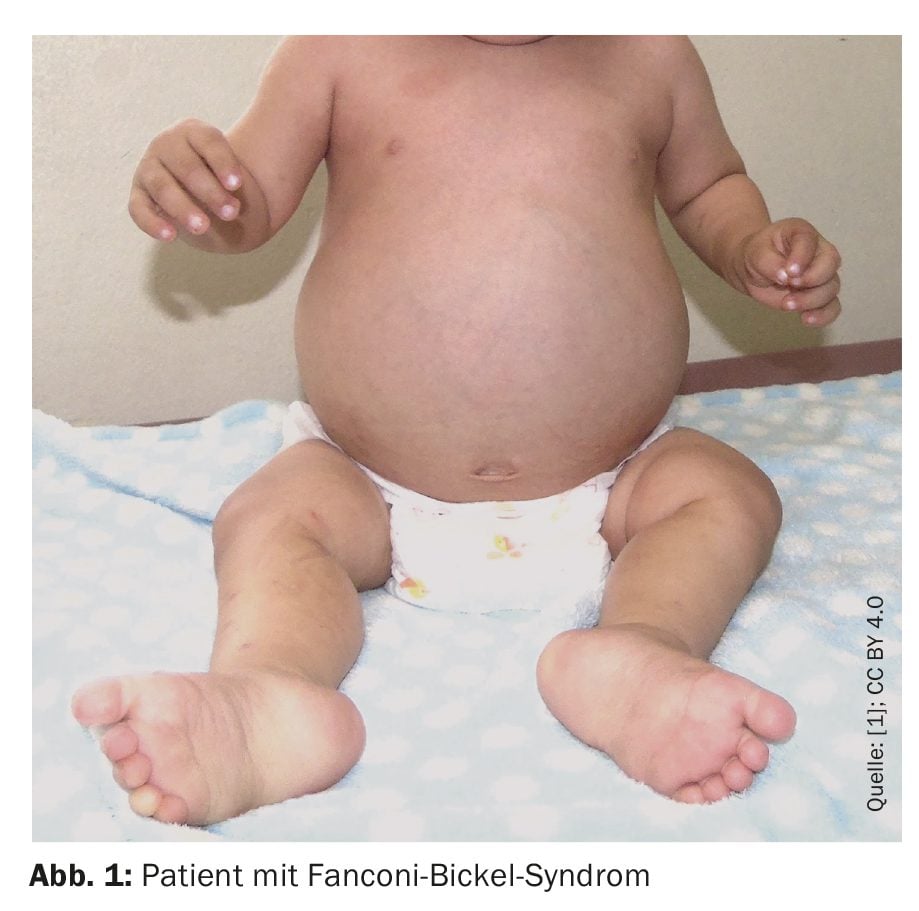
Al ingreso, los médicos diagnosticaron una frente abombada, hepatomegalia, hipotonía y una deformidad pseudomadelungada. El peso y la altura del niño habían estado por debajo del primer percentil desde que tenía seis meses, mientras que su perímetro cefálico estaba dentro de la norma. Las radiografías mostraban deshilachamiento y ensanchamiento de las metáfisis del fémur (distal) y de la tibia (proximal), compatibles con raquitismo.
Los hallazgos clínicos y de laboratorio sugirieron FBS
La joven paciente presentaba hipoglucemia, hipo e hipercalcemia, hipofosfatemia, hipercolesterolemia, hipertrigliceridemia, hiperfosfatemia, hipopotasemia con diarrea y vómitos, hiperlactatemia e hipouricemia. El análisis de orina mostró valores normales de pH y HCO3, pero hiperproteinuria, hipocreatinuria, microalbuminuria, hiperglucosuria e hipercalciuria. El niño también presentaba trombocitosis. La gasometría venosa mostró un pH de 7,261-7,5 mmHg, un HCO3 de 7,5-28,8 mmHg y un exceso de bases de -15,8 a +5,3. Estos análisis llevaron al diagnóstico de acidosis tubular renal. La ecografía abdominal mostró hepatomegalia y no había signos de fibrosis ni nefromegalia.
Una biopsia hepática reveló una arquitectura hepática parcialmente distorsionada debido a la presencia de cierto agrandamiento fibroso del espacio portal, infiltración inflamatoria de linfocitos, hepatocitos grandes y abombados con un patrón en mosaico y fibrosis pericelular leve. La tinción periódica de ácido-Schiff con diátesis pone de manifiesto depósitos eosinofílicos en los hepatocitos que se correlacionan con la deposición de glucógeno. Basándose en estos hallazgos clínicos y de laboratorio, los médicos sospecharon un síndrome de Fanconi-Bickel (FBS).
La secuenciación del exoma identificó una variante patogénica homocigótica
Los resultados de las pruebas genéticas se obtuvieron al año y a los 8 meses de edad utilizando ADN genómico. Se identificaron un total de 131.477 variantes anotadas en 18.179 genes, excluyendo las variantes que probablemente fueran benignas o leves. Para identificar las variantes asociadas al fenotipo de la paciente, se utilizaron los términos “retraso del crecimiento” (HPO: 0001508) y “hepatomegalia” (HPO: 0002240), y los autores señalaron que se consideró un umbral de frecuencia alélica poblacional del 1%. Debido a la hipofosfatemia, el raquitismo y el almacenamiento de glucógeno en el hígado, los investigadores también buscaron manualmente variantes en SLC2A2. Debido a la consanguinidad de los padres, se dio prioridad al análisis de los homocigotos. Se utilizó una frecuencia alélica variante (FAV) superior a 0,9 para seleccionar los posibles genes candidatos. La secuenciación del exoma identificó una variante homocigótica sin sentido en el gen SLC2A2, que se describió como patogénica.
El diagnóstico clínico de la paciente se basó en la presencia de hepatomegalia, hipoglucemia, glucosuria, hipofosfatemia, hiperfosfatemia, hipouricemia, raquitismo, deformidad pseudomadelungada y acidosis tubular renal. Sin embargo, no se pudo determinar la presencia de aminoaciduria, ya que no se disponía de la prueba in situ. Esta excreción insuficiente de aminoácidos habría facilitado el diagnóstico clínico-bioquímico, explican los autores. La confirmación molecular mediante la secuenciación Sanger del gen SLCA2 fue otro obstáculo, ya que esta prueba no está disponible en Perú. Dado el limitado acceso a las pruebas genéticas en el país, no fue posible confirmar la presencia de la variante en los familiares de primer grado de la paciente, subrayan los autores. En este caso, la secuenciación del exoma permitió a los investigadores identificar con precisión la variante homocigota.
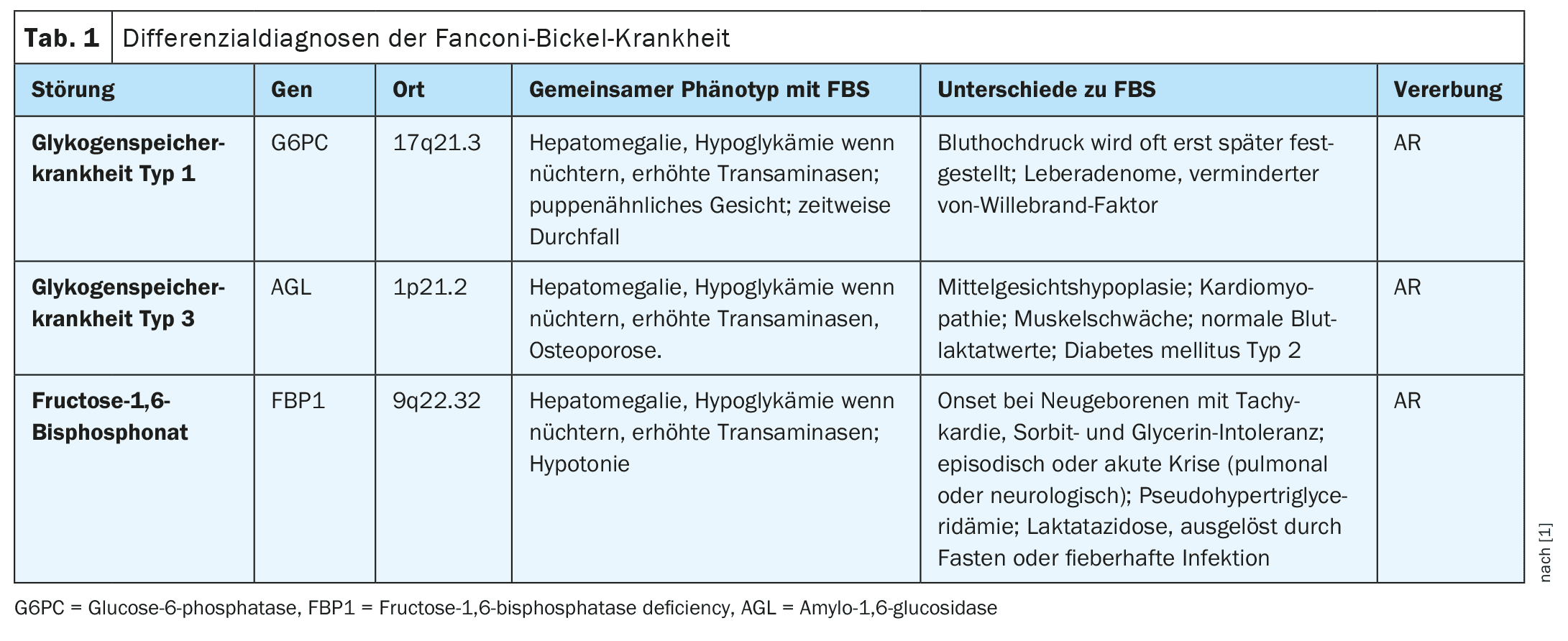
Según el Dr. Abarca-Barriga y sus colegas, el aumento observado del lactato sanguíneo podría deberse a una mayor carga de glucosa procedente del metabolismo anaeróbico. Además, un nivel bajo de ácido úrico debido a la disfunción de los túbulos proximales forma parte del fenotipo de los pacientes con FBS, lo que conduce a la hiperuricosuria. También es probable que algunas características clínicas, como el retraso en el desarrollo del habla, estén asociadas a la presencia de hipoglucemia crónica.
Actualmente no existe ninguna terapia causal para el síndrome de Fanconi-Bickel. La joven paciente fue tratada con bicarbonato, amlodipino, solución de citrato de sodio y ácido cítrico, enalapril, alendronato y zolendronato y recibió tratamiento dietético con almidón de maíz sin cocer, lo que produjo una mejora del peso y la talla. Además, los autores subrayan que la paciente mostró una mejora de 1 desviación estándar (DE) en peso y talla desde el inicio del tratamiento dietético con almidón de maíz sin cocer. Por lo tanto, el uso de almidón de maíz es esencial no sólo para prevenir la hipoglucemia nocturna, sino también para mejorar la talla y el peso del paciente.
Literatura:
- Abarca-Barriga HH, et al.: Importance about use of high-throughput sequencing in pediatric: case report of a patient with Fanconi-Bickel syndrome. BMC Pediatr 2024; 24: 161; doi: 10.1186/s12887-024-04641-1.
HAUSARZT PRAXIS 2024; 19(11): 48–49









